
Case Report | DOI: https://doi.org/10.31579/2690-8794/088
1 MBBS, MS (OBGYN), DM (Medical Genetics), Foetal Medicine & Medical Geneticist, Artemis Hospitals, Gurgaon, India.
2 MBBS, MD (PGI), MRCOG, FRCOG, Diploma in Pelvic Endoscopy, Department of Minimal Access Surgery (Gynaecology), Fortis Memorial Research Institute, Gurgaon.
3 MBBS, MD (Obst & Gynae), Department of Minimal Access Surgery (Gynaecology), Fortis Memorial Research Institute, Gurgaon.
4 MBBS, MS (Obst & Gynae), Department of Obstetrics & Gynecology, Artemis Health Institute, India.
5 MBBS, DNB (Obst & Gynae), MNAMS, Department of Obstetrics & Gynecology, Artemis Health Institute, India.
6 MBBS, MD (Radiodiagnosis), Department of Radiology, Manipal Hospitals, Dwarka, India
*Corresponding Author: Gupta Ashutosh, MBBS, MS (OBGYN), DM (Medical Genetics), Foetal Medicine & Medical Geneticist, Artemis Hospitals, Gurgaon, India.
Citation: Gupta Ashutosh, Aneja Anjila, Bahl Neena, Arora Rupam, Sehgal Renu Raina, Saini Pankaj (2021) Foetal phenotype of Maat-Kievit-Brunner type Ohdo syndrome. Clinical Medical Reviews and Reports. 3(8); DOI: 10.31579/2690-8794/088
Copyright: © 2021, Gupta Ashutosh, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 02 June 2021 | Accepted: 10 September 2021 | Published: 25 September 2021
Keywords: med12; allelic; mkb, ohdo syndrome; micrognathia; cryptorchidism; blepharophimosis; philtrum; vermilion, foetal; maat-kievit-brunner (mkb) type ohdo syndrome; blepharophimosis-mental retardation syndrome; x linked ohdo syndrome
MED12 is a member of large Mediator complex; has a very crucial and central role in RNA polymerase II transcription; regulating cell signals involved in growth, development and differentiation.
Different MED12 mutations may have different clinical presentation representing an allelic disorder.
Maat-Kievit-Brunner (MKB) type Ohdo syndrome; has a typical facial features comprising of blepharophimosis, ptosis, long flat philtrum with thin vermilion, micrognathia with microstomia, scrotal hypoplasia with cryptorchidism, joint hypermobility with clinodactyly with overriding toes,
A primigravida on antenatal ultrasound was detected to have growth restriction, corpus callosal dysgenesis, syndactyly and suspected ambiguous genitalia. Invasive testing and exome sequencing revealed hg19chrX:MED12:c.2315A>G: (p.Lys772Arg);MED12(NM_005120.3):c.2315A>G: (p.Lys772Arg) leading to provisional diagnosis of X linked Ohdo syndrome with an overlap with FG. Missense mutation was classified to be PM2; PP3 (ACMG)
Clinical presentation, phenotype and mutational analysis led to provisional diagnosis of X linked Ohdo syndrome. Maat-Kievit-Brunner type of Ohdo syndrome is a rare condition and this is probably the first case describing foetal phenotype of MKB type of Ohdo syndrome.
In 1974, Opitz and Kaveggia described a X-linked recessive intellectual disability syndrome characterized by relative macrocephaly, hypertelorism, downslant palpebral fissures, prominent forehead with frontal hair upsweep with broad thumbs and halluces [1].
In 1984, Lujan et al reported four male patients with marfanoid habitus, long narrow face, small mandible with high-arched palate, hypernasal voice with intellectual disability [2].
In 1993, Maat-Kievit et al delineated a X-linked Ohdo syndrome; characterized by failure to thrive with facial features comprising blepharophimosis, ptosis, wide depressed nasal bridge, long philtrum, thin vermilion, micrognathia with microstomia, scrotum hypoplasia with cryptorchidism, joint hypermobility, overriding toes with clinodactyly, cafe-au-lait spots, developmental delay with hearing disability [3].
In 2007, Risheg et al documented a recurrent MED12 mutation (p.Arg961Trp) which was earlier diagnosed as FG (Opitz–Kaveggia) syndrome [5]. Reported a different mutation (p.Gly958Glu) with similar clinical phenotype [4]. Demonstrated (p.Asn1007Ser) MED12 mutation in a family originally diagnosed as Lujan–Fryns syndrome [6].
Vulto-van Silfhout [11] reported (p.Arg1148His, p.Ser1165Pro and p.His1729Asn) mutations in a family originally documented as MKB. [7] Isidor et al. identified (p.Arg1148His) mutation in two male siblings with phenotype similar to those of MKB [8].
It is now established that MED12 mutations have different clinical presentation representing an allelic disorder. (Table 1) [9].

Maat-Kievit-Brunner (MKB) type Ohdo syndrome has a X-linked recessive inheritance pattern with typical facial features like thick alae nasi, blepharophimosis, ptosis, wide depressed nasal bridge, large bulbous nose, long flat philtrum with thin vermilion, micrognathia with microstomia, feeding difficulties, scrotal hypoplasia with cryptorchidism, joint hypermobility with clinodactyly with overriding toes, cafe-au-lait spots, hearing disability. Facial features become more pronounced with age with characteristic triangular facies [1]. It may have mild to severe developmental delay with delayed motor milestones with behavioural problems with mental retardation.
Maat-Kievit-Brunner type of Ohdo syndrome is a rare condition and this is probably the first case describing foetal phenotype of MKB type of Ohdo syndrome. It is caused MED12 gene (mediator complex subunit 12) mutations; which is a subunit of the mediator complex, a group of about 25 proteins which regulates gene activity. It physically links transcription factors to turn on or off the genes. MED12 protein is involved in development of the neurons, in several chemical signalling pathways to regulate cell growth, migration and differentiation.
A booked primigravida at 19 weeks of pregnancy with no high risk factor was evaluated in the department of Foetal Medicine at Artemis Hospital for foetal anomaly scan. It is a non-consanguineous marriage and no other male member of the extended family has been affected with intellectual disability or dysmorphism.
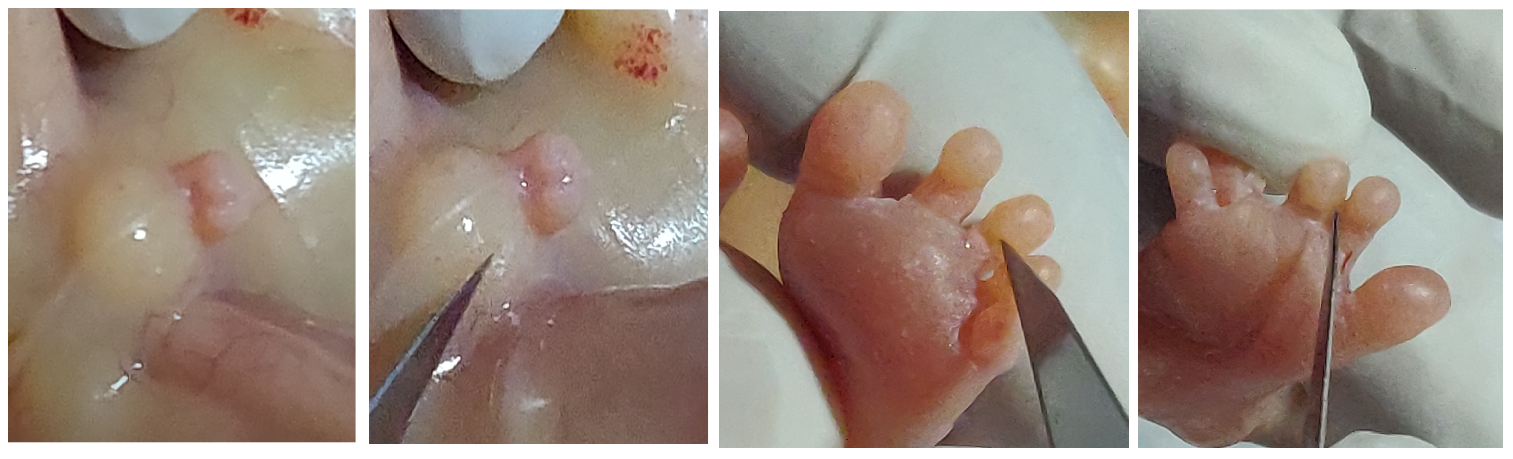
On antenatal ultrasound, high riding / upward displacement of 3rd ventricle was noticed with divergent frontal horns suggestive of corpus callosal dysgenesis (Figure 1-a). Further evaluation showed syndactly of 2 and 3rd toe of the right foot; however 2 and 3rd toes of the left foot were seen separate (Figure 1-b). Foetus was also identified to have hypospadias with suspected sex reversal or ambiguous genitalia (Fig 1-c). There was also an element of growth restriction by 1 week at 19 weeks of gestation (Figure 1-d).
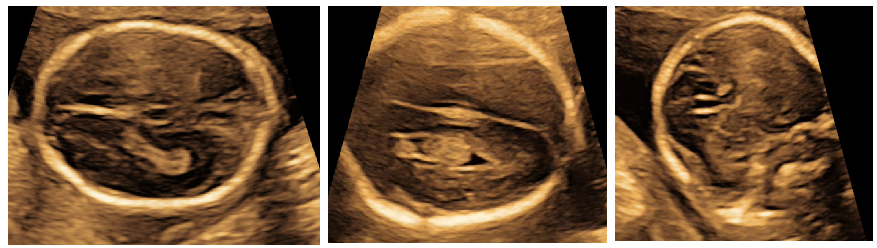


Axial scan of foetal brain showing high riding 3rd ventricle; upward displacement of the third ventricle; increased separation of the hemispheres, Coronal scan of a normal foetal brain demonstrating absence of the corpus callosum (genu), an increased distance between the frontal horns and their abnormal aspect suggestive of corpus callosal dysgenesis (a)
In the advent of foetal growth restriction, corpus callosal dysgenesis, syndactyly and suspected ambiguous genitalia; the consult and (parents) was counselled for invasive foetal testing; amniocentesis was done and on exclusion of maternal cell contamination; foetal DNA was processed for exome sequencing. Ambiguous genitalia in the clinical picture prompted and pointed towards foetal cholesterol pathway defect thus an exome sequencing was ordered. With all this information, possibilities couple decided for an irreversible decision regarding the pregnancy.
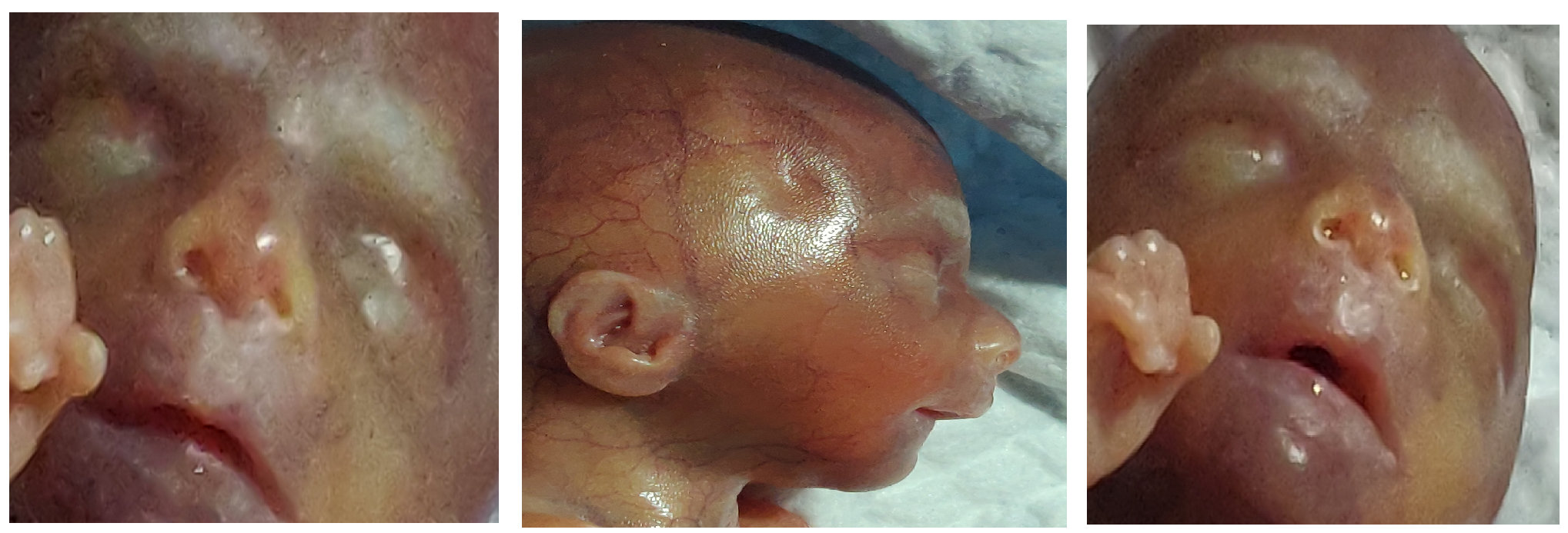
Postnatal examination of the foetus showed penoscrotal hypospadias, syndactyly in 2&3rd toe in the right feet and normally separated 2 and 3rd toe in the left toe (Figure 2-a) corroborating the main antenatal findings. It also revealed bilateral low set ears, micrognathia (Figure 2-b), thin vermilion, long philtrum and blepharophimosis (bilateral and both eye lids could not be separated completely with considerable effort) (Figure 2-c) corroborating the diagnosis of X linked Ohdo syndrome. The consult and (parents) gently refused for further postnatal (post mortem) examination of the foetus.

Exome sequencing revealed hemizygous missense variation single-nucleotide variant (SNV) in exon 16 of the MED12 gene (Xq13.1); with amino acid substitution of Arginine for Lysine at codon 772, c.2315A>G (p.Lys772Arg); hg19chrX:MED12:c.2315A>G:(p.Lys772Arg); MED12(NM_005120.3):c.2315A>G:(p.Lys772Arg) leading to provisional diagnosis of possible MED12 related disorder; X linked Ohdo syndrome or FG syndrome.
The same mutation was confirmed by sanger sequencing and identified in the asymptomatic mother. In silico prediction of the variant has been identified to be damaging by LRT and Mutaion Taster.
In accordance to the American College of Medical Genetics and Genomics (ACMG) criteria for classifying pathogenic variants; the variation was classified as variant of uncertain significance and the missense mutation was classified to be PM2 (absent from controls or at extremely low frequency if recessive) and PP3 (multiple lines of computational evidence support a deleterious effect on the gene or gene product). (19)
Table no 1 elaborates characteristic features of the Ohdo syndrome; some of which detected antenatally were confirmed and remaining were identified by postnatal examination. Maat-Kievit et al. (1993), Verloes et al. (2006) and Vulto-van Silfhout et al. (2013) (3, 7, 14) have reported missense mutations in MED 12 to be causative for Maat-Kievit-Brunner type Ohdo syndrome (Table 2). Table no 3 describes the genotype – phenotype corelation of MED 12 mutations.


Clinical presentation, phenotype and mutational analysis led to provisional diagnosis of possible MED12 related disorder; X linked Ohdo syndrome or FG syndrome.
MED12 being a member of large Mediator complex; has a very crucial and central role in RNA polymerase II transcription. It regulates the cell signals involved in growth, development and differentiation.
MKB is a rare condition; X-linked recessive inheritance; very typical pattern of facial features which are difficult to be detected on antenatally. It is probably the first case of detection of foetal phenotype of MKB.
With ever increasing use of massive parallel sequencing with next-generation sequencers, it is likely that more MED12 distinctive phenotype and mutations will be detected. This also helps in identifying the genetic aetiology in families with intellectual disability and dysmorphic features and to broaden genotype–phenotype correlations.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
