
Review Article | DOI: https://doi.org/10.31579/2766-2314/060
Department of Chemical and Biomolecular Engineering, National University of Singapore.
*Corresponding Author: Wenfa Ng, Department of Chemical and Biomolecular Engineering, National University of Singapore.
Citation: Wenfa Ng (2021) Evaluating the Potential of Applying Machine Learning Tools to Metabolic Pathway Optimization. J, Biotechnology and Bioprocessing 2(9); DOI: 10.31579/2766-2314/060
Copyright: © 2021, Wenfa Ng, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 05 October 2021 | Accepted: 18 October 2021 | Published: 28 October 2021
Keywords: pathway optimization, machine learning tools, enzyme activity prediction, promoter classification, expression tuning
Successful engineering of a microbial host for efficient production of a target product from a given substrate can be viewed as an extensive optimization task. Such a task involves the selection of high activity enzymes as well as their gene expression regulatory control elements (i.e., promoters and ribosome binding sites). Finally, there is also the need to tune expression of multiple genes along a heterologous pathway to relieve constraints from rate-limiting step and help reduce metabolic burden on cells from unnecessary over-expression of high activity enzymes. While the aforementioned tasks could be performed through combinatorial experiments, such an approach incurs significant cost, time and effort, which is a handicap that can be relieved by application of modern machine learning tools. Such tools could attempt to predict high activity enzymes from sequence, but they are currently most usefully applied in classifying strong promoters from weaker ones as well as combinatorial tuning of expression of multiple genes. This perspective reviews the application of machine learning tools to aid metabolic pathway optimization through identifying challenges in metabolic engineering that could be overcome with the help of machine learning tools.

Metabolic engineering sought to increase the production of small molecules using cellular metabolism that has been rewired through genetic engineering. Comprising a workflow that can be categorized into design-build-test-learn cycle, a typical metabolic engineering project would involve iterative design and test experiments aiming to improve the expression of desired genes and production of target metabolites. As such, many aspects of engineering a microbe for overproducing a metabolite can be characterized as an optimization problem. Specifically, a key concern in metabolic engineering has been the selection of enzymes with high activity under a broad range of conditions, and strong promoters and ribosome binding sites, and the tuning of expression of multiple genes [1-4]. These goals could not be achieved through rational selection, but only with trial and error experimentation. The latter is not desirable considering the time and effort involved, and this has sowed the seeds for introducing data-driven algorithmic approaches to the traditional confines of metabolic engineering.
Although optimization can be performed by various algorithms, the latest trend has been the application of machine learning tools in optimization problems. In general, machine learning algorithms sought to identify patterns hidden in large datasets, and this enabling feature has been used in different aspects of metabolic engineering such as pathway optimization. Fundamentally, machine learning algorithms builds a model from input data using an iterative cycle of parameter fitting to a curvilinear description of the data. The obtained machine learning model could subsequently be used in predicting properties of enzymes or pathways using certain inputs. This thus arrives at the core enabling feature of machine learning: it is an automated search for a set of mathematic descriptions that describe particular sets of data. Usually thought to be data intensive, machine learning tools could also be applied to small datasets with or without data augmentation [5-7], and this latter feature dovetails with the inherently small scale nature of many biological datasets.
Thus far, machine learning has been applied to many but not all aspects of metabolic engineering and pathway optimization. For example, machine learning has been utilised in reconstruction of metabolic model of a species [8-12]. De novo pathway engineering is another aspect that has benefited from application of machine learning tools [13]. Machine learning tools have also enabled the deciphering of kinetic parameters of enzymes from metabolomics data [14,15]. In the same vein, correlations between expression level and design of various gene expression control elements (e.g., promoter and ribosome binding site) has been sought using the tools of machine learning [16].
This article sought to review areas where machine learning has informed pathway optimization. These include: (i) selecting enzymes with the highest activity for a pathway, (ii) selecting promoters and ribosome binding site of appropriate strength for particular genes, and (iii) tuning the expression of multiple genes in a pathway (Figure 1). But, the journey marched by machine learning in metabolic pathway optimization remains incomplete. For example, opportunities exist in applying machine learning to predict the regulatory motifs of enzymes and pathway dynamics from multiomics data [17], as well as assessing the performance of microbial cell factory [18].

Essentially, a metabolic pathway comprises a set of reactions that transform a substrate into a product through a series of bond formation and cleavage. Both expert knowledge and retrobiosynthetic approaches could be used in developing this pathway [11]. In particular, machine learning methodologies have been successfully applied in retrobiosynthesis and is reviewed elsewhere [11, 19]. With a set of coupled sequential reactions in mind, the next step is in selecting the appropriate enzymes for performing the respective reactions. Recently, machine learning has helped refine gene annotation through better recognition of genomic signals such as polyadenylation signals and translational start site [20]. In particular, deep learning approaches have played an important role in dissecting the often convoluted signals from the genome in assigning gene function to sequence information [21-25], and is poised to help identify more enzyme candidates with suitable functions in a metabolic engineering project. The latter comes about due to enzyme promiscuity where some enzymes could be repurposed for other functions [26,27]. Usually, enzymes with the highest activities and performance are desired. But, given the plethora of similar enzymes in different species, how does one select the best performing enzyme for an application? Can machine learning help rule out some candidates that are unlikely to work? In addition, which performance measure should be the basis for optimization? Since enzyme performance can be described by turnover number, inhibitory concentration (Ki) and binding affinity between substrate and enzyme (Km), multiple parameters could be used in machine learning tasks for predicting enzyme performance with amino acid sequence as input. But, the challenge lies in relative lack of full set of characterization data for different enzymes in varied species. Such data are incomplete given the effort and resources needed to perform detailed biochemical assays for each substrate.
One example of applying machine learning to predicting enzyme activity is in using an ensemble of enzyme characteristics such as biochemical parameters and structure to inform enzyme catalytic turnover number, which is a proxy parameter for enzyme activity [28] (Figure 2). Correlations between catalytic turnover number and enzyme structure elucidated by the machine learning tools hold important implications for how structural biology could inform enzyme biochemistry [28]. Indeed, other studies have also corroborated that enzyme conformation can be reliably correlated with enzyme activity level [29,30]. Furthermore, it has been shown that sequence alone could not accurately describe enzyme activity [31]. However, a study has shown that combination of sequence information and structural descriptors of enzyme-substrate recognition is useful for predicting enzyme function and activity [31]. Hence, the current state-of-the-art in enzyme activity prediction remains firmly in the realm of structure-activity correlate, with attempts at extending the correlation to the sequence level meeting challenges at our inability to resolve the protein folding problem. But future advances in using machine learning to circumvent the protein structure prediction problem may ultimately tie the link between enzyme sequence and activity level.

Optimization of gene expression regulatory elements
Gene expression regulatory elements such as promoters and ribosome binding site (RBS) controls the level at which the heterologous genes could be expressed. To facilitate selection of appropriate promoter and RBS for tuning the expression of heterologous genes, a need exists to build a predictive model able to correlate promoter or RBS sequence with expression level.
Theoretically, building a machine learning model capable of predicting promoter or RBS strength from sequence information does not necessarily require accurate definition of promoter or RBS sequence, which remains a research topic [32]. But, if promoter sequences in a training dataset are accurate, this would reduce the noise in the model and afford more accurate prediction. Hence, the computational challenge in applying machine learning to promoter strength prediction lies in the identification of small snippets of nucleotide sequence that strongly correlates with expression level [33]. Currently, a commonly used method for extracting sequence features is position weight matrix [34], but the approach may not be transferable to different species [33]. Another problem with promoter strength prediction is the relative lack of data, particularly in cases where machine learning is applied to experimentally characterized promoters [35,36]. But, use of genome-wide RNA-seq data may provide sufficient data that significantly improves machine learning based predictions of promoter strength.
Typically, the input data for training are promoter or RBS sequence and expression level as measured by protein or mRNA transcripts abundance. Such data could be modelled by support vector machine algorithms [36,37] (Figure 3), but recently, deep learning methods have also been applied to the problem and have shown promising results [38,39]. One approach uses pseudo-dinucleotide composition coupled to CNN for both promoter identification and strength prediction in prokaryotic organisms [39]. The method demonstrated good performance compared to state-of-the-art methods, but it is still limited to classifying promoters into strong or weak promoters, which does not provide metabolic engineers with the ability to achieve fine-grained control over gene expression. Another approach took into consideration evolutionarily relationships between orthologous genes and showed that such a methodology provided better predictions of mRNA abundance from DNA sequence [40]. Overall, neural network-based approaches may not be the only way forward in promoter strength prediction, particularly in cases with small datasets. For example, kernel-based approaches such as support vector machines have provided better performance compared to artificial neural network in some instances [36].

In comparison to promoters, ribosome binding sites are more well-defined. This comes about due to the structure of gene regulatory region where ribosome binding sites (RBS) are downstream of the transcriptional start site (TSS), which could be experimentally defined by RNA-seq data [33]. Similar to promoters, RBS are important modulators of gene expression level given that it governs the strength of binding between the small subunit (SSU) of ribosome with the mRNA transcript obtained after transcription. In a recent study, machine learning tools were used in defining the RBS sequence-phenotype relationship, which forms the basis for predicting optimal RBS sequences for multi-gene pathway. Computational predictions were validated through experiments and demonstrated the approach’s utility in enabling screening of a large combination of RBS sequences for multi-gene pathway [41]. But, in general, correlation between RBS sequence and expression level may not be easily discernible by machine learning tools. For example, a recent study did not find strong correlation between experimental protein expression data and predicted RBS strength [42], thereby, indicating room for improvement in the application of machine learning to RBS strength prediction. One major hurdle in RBS strength prediction comes from the relatively small sequence space of these gene regulatory elements as RBS are inherently shorter than promoters. Lack of sufficient variability in expression level from the small RBS sequence set would thus severely hamper prediction of protein expression level from RBS sequence.
Expression of a heterologous gene in a cell incurs a metabolic burden. For long pathway comprising multiple genes, such metabolic burden may have a detrimental effect on cell growth. In other situations, there may be excessive expression of enzyme for a particular step of the pathway that may lead to depletion of an intermediate metabolite needed to maintain other critical pathways of the cell. Hence, a need exists in tuning the expression of individual gene in a pathway to ensure that only sufficient enzymes are expressed to enable proper functioning of the pathway and deliver higher yields, and preventing metabolic choke points from emerging. Tools available for tuning the expression of multiple genes in a pathway would be promoter and ribosome binding site. Since heterologous genes are typically expressed in an operon in prokaryotic hosts, ribosome binding site tuning are more often used in prokaryotes. On the other hand, need for individual promoter for each gene of the pathway in eukaryotic hosts meant that promoter tuning is as important as ribosome binding site tuning in eukaryotes. Combinatorial tuning of promoter and RBS may thus afford fine-grained control over gene expression in eukaryotes.
Conceptually, the problem of optimizing expression of individual genes in a pathway can be depicted as a search for optimal levels of individual enzyme in a gene expression landscape. Statistical design of experiments approach has been put to use in this endeavour, yielding promising results that reduce experiment effort [43,44]. However, such search for the optimal combination of promoter and RBS usually will not arrive at the global optimum. In particular, extent in which the gene expression landscape is sampled determines the likelihood in which an optimal could be obtained. As the number of tunable parameters (promoter and RBS) increases with each additional gene in the pathway, the optimization problem could quickly escalate in complexity and size beyond the search capability of conventional optimization algorithms.
One way to circumvent the problem is through employing machine learning to detect hidden mathematical relationships between different sampled points on the gene expression landscape where combinatorial pathway optimization experimentation help deliver the data points that feeds the machine learning algorithms (Figure 4). In a recent example, artificial neural networks are employed to glean relationships between product titer of different strains with different promoters in a combinatorial optimization exercise. Predictions from the machine learning algorithms were verified experimentally, thereby, demonstrating the utility of the approach [45]. Besides neural networks, support vector machines and other kernel-based approaches may also be useful for such applications. However, how well machine learning performs critically depends on the characteristics of the input dataset and its size. Better predictions would naturally come from a larger dataset, which places greater demand on experimentation in combinatorial tuning of expression of multiple genes. In addition, input data should also cover a wide range in order to achieve a large dynamic range for corresponding predictions of product yield, titer, and productivity. At a more fundamental level, much room exists for the utilization of machine learning approaches in combinatorial pathway optimization since most studies in the field still relies on statistical design of experiment or construction of smart libraries to expedite search for optimal gene expression level of a pathway [46,47]. Developing better methods to efficiently and cost-effectively generate the input data for training various machine learning models remain an important research topic.
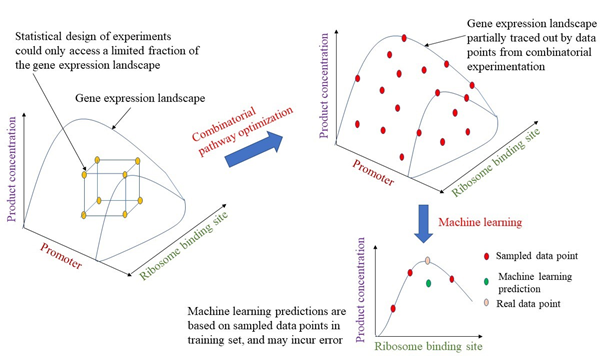
Machine learning has been applied to many facets of metabolic engineering and pathway optimization. From selection of enzymes to tuning of gene regulatory elements, machine learning’s greatest strength has been the gleaning of hidden patterns in complex data set to help offer solutions in new situations through building a predictive mathematical model. Such automated tools significantly ease the burden on metabolic engineers in making critical decisions such as gene selection and promoter choice during pathway optimization. But, application of machine learning tools to metabolic engineering remain a significant challenge for the novice researcher. This is made even harder by the cryptic nature of machine learning algorithms. Thus, more resources may be provided to enable researchers to begin integrating these tools across the pathway development process.
The author declares no conflicts of interest.
No funding was used in this work.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
