
Research Article | DOI: https://doi.org/10.31579/2690-4861/307
Department of Thoracic Surgery, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, PR China.
*Corresponding Author: Yu Liu, Department of Thoracic Surgery, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, PR China.
Citation: Zhang W., Zhu W., Xu S., Liu Y., (2023), Establishment and Validation of Prognostic Model of Lung Adenocarcinoma Based on Apoptosis-Related Genes, International Journal of Clinical Case Reports and Reviews, 13(5); DOI: 10.31579/2690-4861/307
Copyright: © 2023, Yu Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 10 April 2023 | Accepted: 25 April 2023 | Published: 02 May 2023
Keywords: apoptosis; lung adenocarcinoma; prognosis; risk model; biology message
Objective: Apoptosis is an autonomous cell death process controlled by genes that can keep the internal environment of cells relatively stable. Lately, many studies have shown that apoptosis plays an important role in the construction of tumor microenvironment, mainly through the release of a series of regulatory factors to control cell growth. Therefore, it is very valuable to find and explore the possible role of apoptosis in the pathogenesis of lung cancer and its impact on the prognosis of patients.
Materials and Methods: We selected 10 apoptosis-related genes and used different statistical methods to construct a model to predict the survival and prognosis of patients with lung adenocarcinoma. Univariate and multivariate Cox analysis were used to evaluate the predictive power and value of prognostic models, as well as receiver operating characteristic (ROC) curve analysis.
Results: We defined two subgroups (cluster1 and cluster2). Compared with cluster1, cluster2 was named "apoptosis inhibition subgroup". The survival prognosis of cluster1 was significantly better than that of cluster2, and the infiltration of immune cells in cluster2 suggested that its immune system was inhibited. Then, we established a new prognostic model of apoptosis-related genes to identify high-risk patients with lung adenocarcinoma. Two types of Cox proportional hazards analysis and ROC curve showed that our model can be used as a new independent factor for the prognosis of lung adenocarcinoma patients, and had strong predictive ability.
Conclusion: Apoptosis is involved in the formation and development of lung adenocarcinoma and plays an important role. Our survival model can better predict the survival and prognosis of lung adenocarcinoma, and our study may be helpful for further research on new therapeutic targets and precise individualized treatment in the future.
Lung cancer is a malignant tumor with high incidence rate and high mortality in worldwide[1, 2]. The incidence rate of non-small cell lung cancer (NSCLC) accounts for about 85% of the total incidence of lung cancer, and lung adenocarcinoma (LUAD) accounts for 40% [3]. Although considerable progress has been made in exploring lung cancer biology and multimodal treatment, prognosis of lung cancer patients is still far from satisfactory. This is partly because the current guided staging method could not accurately predict the prognosis of lung cancer. The reason why some patients with early lung cancer relapse or metastasize is that they do not receive effective adjuvant therapy after operation [4, 5]. Through some current diagnostic methods, such as computed tomography, about 40% of lung cancer patients are still diagnosed with distant metastasis [6]. Therefore, finding reliable predictors of prognosis will bring great value to guide the treatment of LUAD [7].
Apoptosis is a special biological process, which leads to pathological changes different from necrosis by activating an evolutionarily conserved intracellular pathway, and finally leads to programmed cell death. Apoptosis involves several biological and pathological processes, such as embryonic development, steady-state maintenance of tissues and organs, tumorigenesis and tumor progression [8, 9]. At present, the existing research shows that apoptosis is involved in the process of tumor occurrence and development, and has an important impact on tumor treatment [10]. As one of the characteristics of cancer, acquired resistance to apoptotic cell death is mainly due to the overexpression of anti-apoptotic genes and the down-regulation or mutation of Pro apoptotic related genes [11]. At present, there is no method based on apoptosis-related genes expression to identify high-risk patients with LUAD.
This study screened hallmark gene sets enriched in the apoptotic pathway from the Molecular Signatures Database(http://www.gsea-msigdb.org/gsea/index.jsp),and constructed the co-expression network of apoptosis-related genes in LUAD. Using Cox analysis, we selected the expression characteristics of the first 21 apoptosis-related genes with HR significance to characterize the different apoptosis states of LUAD samples, and identified apoptosis status in the characteristics of LUAD patients as the main risk factor for overall survival. Then we studied an independent cohort to verify its prognostic value. In addition, this paper also describes the infiltration of 22 immune cell types in LUAD, the clinical characteristics of patients, and their correlation with the different expression of apoptosis-related genes. These results are helpful to further prove that our model has better prognostic ability, and to further explore and reveal the mechanism of apoptosis in the development of LUAD.
Source of sample data
In our study, the gene expression data and clinical data of lung cancer patients were collected from The Cancer Genome Atlas (TCGA) database (https://tcga-data.nci.nih.gov/tcga/). After excluding patients with repeated and missing clinical information, we analyzed the data of the remaining 494 patients. Among them, 248 patient data were used as the training set, the remaining 246 patient data were used as the validation set. Through the different analysis, there was no significant difference in age, gender, stage and other clinical characteristics between the two groups (Table 1). All gene expression data in this study were standardized, and we only used open-access data, which meets the requirements of ethical review.
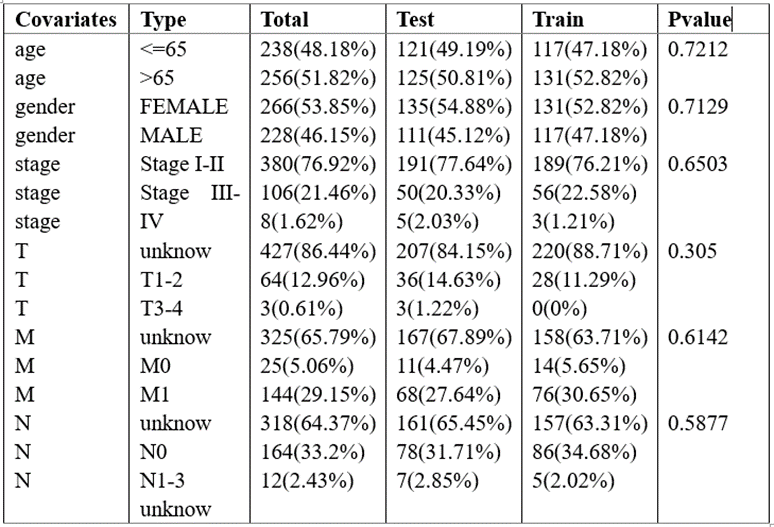
Table 1: There was no significant difference in clinical characteristics between training set and validation set.
Screening and obtaining of apoptosis-related genes
A list of transcriptome profiling data of apoptosis-related gene and hallmark gene sets was obtained from the Molecular Signatures Database (MSigDB) (http://www.gsea-msigdb.org/gsea/index.jsp). A total of 1434 apoptosis-related genes were screened out, and finally we obtained 21 the most significant genes through Cox analysis.
Correlation between immune characterization and apoptosis
We obtained data on 22 immune cells, including T cells CD8, T cells CD4 naive, T cells CD4 memory activated, T cells CD4 memory resting, T cells follicular helper, T cells regulatory (Tregs), T cells gamma delta, B cells naive, B cells memory, Monocytes, NK cells resting, NK cells activated, Plasma cells, Macrophages M0, Macrophages M1, Macrophages M2, Dendritic cells resting, Dendritic cells activated, Mast cells resting, Mast cells activated, Eosinophils and Neutrophils. All data are downloaded from the ImmuCellAI database. We used limma software package to analyze the relationship between the infiltration of these immune cells and the expression level of apoptotic-related genes. And then we assessed the infiltration difference of them.
Identification of apoptosis-related genes prognostic signatures for LUAD
We identified 21 apoptosis-related genes by using univariate Cox proportional hazard analysis. After that, the least absolute shrinkage and selection operator (LASSO) Cox regression model was used to further screen for the best prognostic markers with P<0>
Risk score = coef (gene1) × expr (gene1) +
coef (gene2) × expr (gene2) +
……
coef (gene n) × expr (gene n).
coef (gene n) is the coefficient of each gene.
Expr (gene n) is defined as the expression of apoptosis-related genes.
n represents the number of apoptosis-related genes selected in our model.
It means that the gene plays a protective role when the coefficient of gene is less than 0, and if it is greater than 0, it means that the prognosis of patients will become worse. According to the median risk score, we divided the patients into two groups: high-risk and low-risk group, and evaluated the difference between them. Then we used the survival ROC R package to assess the accuracy of the prediction function of the model. In addition, we also evaluated the relationship between risk score (RS) and clinical factors.
Results
Consensus Clustering Showed Different expression of apoptosis related-genes in the Two LUAD Gene Clusters
A total of 1434 apoptosis-related genes were showed in the Molecular Signatures Database (MSigDB), and the PPI network is shown as Figure 1A. Then we screened and analyzed the expression characteristics of a group of related genes highly enriched in apoptosis related pathways. In order to understand their role in LUAD, we studied the differences of these biological signals in tumor tissues and normal tissues, and found that many genes expressed abnormally in the LUAD samples, as shown as Figure 1B. And we selected 21 genes including 4 down-regulated and 17 up-regulated genes with significant differences (Figure 1C).
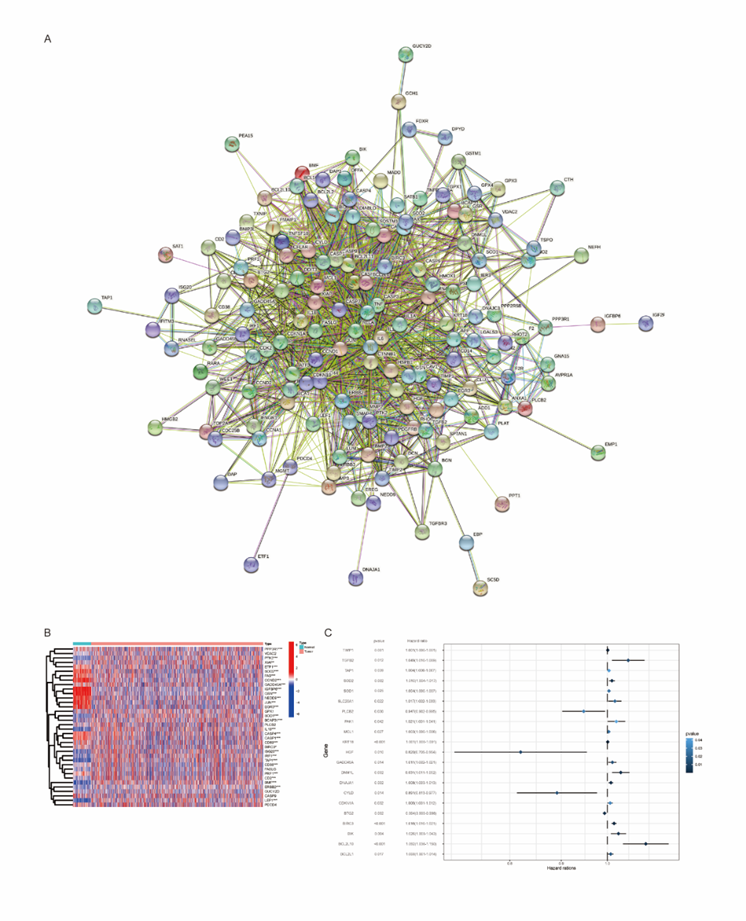
Figure 1: 21 genes were found to be significantly associated with apoptosis.
Then, we used the Consensus Clustering Method to cluster 494 TCGA LUAD tumor samples to determine their apoptotic status. The result demonstrated that when the value of K is 2, the interference between subgroups is smaller than that in other cases (Figure 2A). Therefore, we finally identified them as two subgroups, namely cluster1 (n=416) and cluster2 (n=78). We compared the transcriptional profiles between cluster1 and cluster2 by principal component analysis, and found that there were significant differences between two subgroups (Figure 2B). The expression level of apoptosis-related genes was higher in cluster1, but lower in cluster2, so we defined the cluster2 as “apoptosis inhibition subgroup” compared with cluster1. Through Kaplan-Meier survival analysis, we found that cluster1 with high expression of apoptosis-related genes has better overall survival (OS) than cluster, P < 0>Figure 2C).
Immune Cells Infiltration of Different apoptosis-related gene expression in LUAD
Some evidence suggested that immune cells infiltration of tumor was associated to clinical response of treatment and cancer prognosis, including LUAD. Therefore, we analyzed 22 kinds of immune cells in LUAD, including 7 T-cell subtypes, 2 B-cell subtypes and Monocytes, NK cells resting, NK cells activated, Plasma cells, Macrophages M0, Macrophages M1, Macrophages M2, Dendritic cells resting, Dendritic cells activated, Mast cells resting, Mast cells activated, Eosinophils and Neutrophils. Figure 2D shows the different infiltration of immune cells in the two subgroups.
Apparently, the Dendritic cells and Mast cells that may play a promoting role in killing tumor cells in cluster2 have less infiltration, while Tregs and macrophages are more, while cluster1 shows the opposite trend. The results showed that the immune system in cluster2 were inhibited, which predicted the poor survival prognosis (Figure 2E).
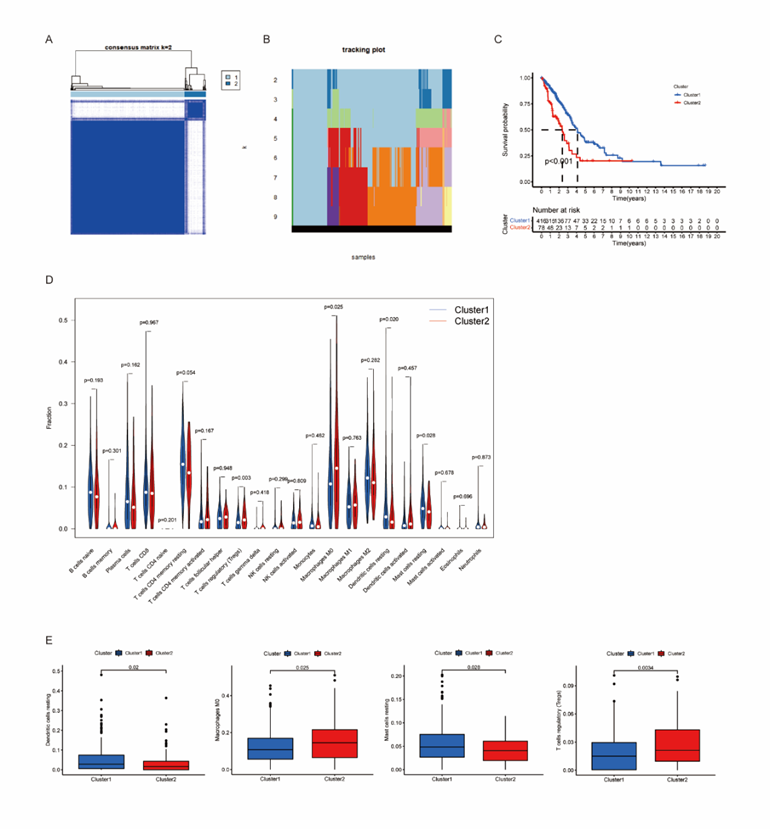
Figure 2: Consensus Clustering identified two clusters of LUAD with different apoptosis status.
Establishment of prognostic model of apoptosis-related genes
In order to find prognostic indicators related to apoptosis, we performed Cox regression analysis on different gene expression and overall survival (OS) data of 248 LUAD samples. The results showed that 21 genes were significantly associated with OS, of which 17 genes were highly expressed and the other 4 genes were low expressed. The results of lasso Cox regression analysis (Figure 3A, B) showed that 9 up-regulated genes (DNM1L, SOD1, TGFB2, BIRC3, KRT18, BIK, DNAJA1, TIMP1, BCL2L10) and one down-regulated gene (CYLD) may be the most powerful prognostic markers.
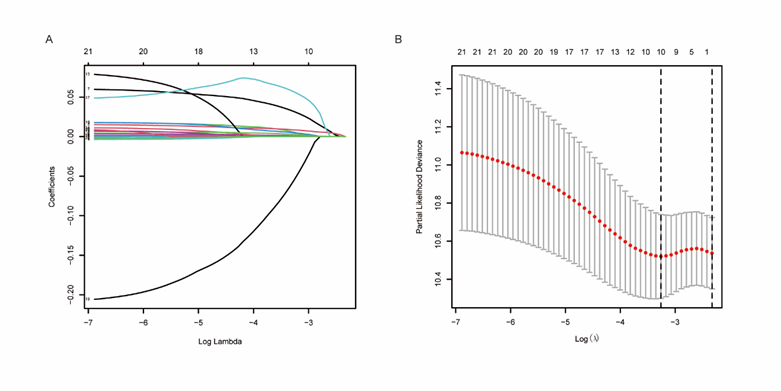
Figure 3: Establishment of apoptosis related gene markers.
(A, B) Lasso Cox regression was used to construct effective prognostic markers.
The coefficients of 10 genes are shown in Table 2. We used Lasso analysis to calculate the coefficient of each marker to obtain the risk score calculation formula, as shown below: risk score = 0.005534 * expr (DNM1L) + 0.001061 * expr (SOD1) + 0.031586 * expr (TGFB2) + 0.008554 * expr (BIRC3) + 0.000488 * expr (KRT18) + 0.004843 * expr (BIK) + 0.002794 * expr (DNAJA1) + 0.000655 * expr (TIMP1) + 0.056489 * expr (BCL2L10) + (-0.054429) * expr (CYLD). All samples were scored by the above formula and divided into high-risk group and low-risk group according to the median. And the results of Kaplan-Meier analysis showed that compared with the low-risk group, the prognosis of the high-risk group was significantly worse (Figure 4A).
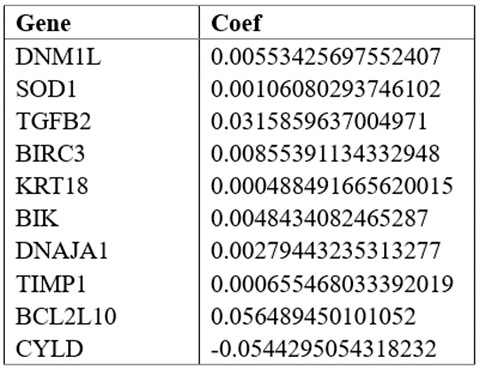
Then, we evaluated the model and survival status of LUAD patients and visualized them by using risk curve and scatter plot (Figure 4B-D). This indicated that the occurrence of mortality depends on the risk score. The heatmap of these 10 apoptosis-related genes showed, 9 genes (DNM1L, SOD1, TGFB2, BIRC3, KRT18, BIK, DNAJA1, TIMP1, BCL2L10) were highly expressed in high-risk group, while CYLD was highly expressed in low-risk group.
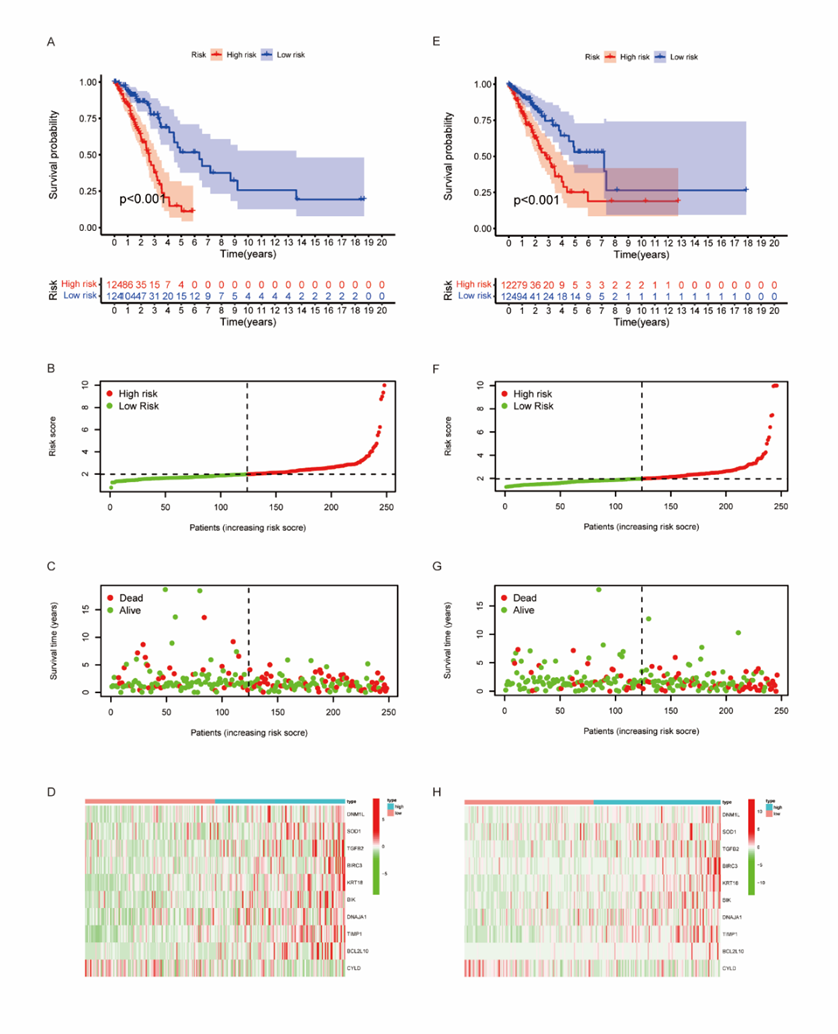
Figure 4: Establishment of prognostic risk scoring model.
Evaluation and Validation of the survival model for LUAD patients
We performed univariate and multivariate Cox regression analysis to evaluate whether the survival model of these 10 apoptosis-related genes can be used as independent prognostic factors of LUAD. In univariate Cox regression analysis, we found that tumor stage was related to OS, as well as risk score. The hazard ratio (HR) of risk score was 1.363(95% CI 1.229-1.511) (P < 0>Figure 5A, B).
The area under ROC (AUC) curve was used in TCGA train set and test set to evaluate the sensitivity and specificity of the risk scoring model in 1-, 3- and 5-year prediction. We calculated the area under the ROC curve of the risk score, and the results showed that the model has a good effect in predicting the prognosis (Figure 5C).
We calculated the risk score from 246 samples in the test set and validated our model. Based on the above formula and the best dividing point, we calculated the risk score of each sample and divided these patients into high-risk group (n=122) and low-risk group (n=124). It should be noted that, consistent with the results of the train set samples, the prognosis of the high-risk group was significantly worse in the test set (P < 0>Figure 4E). The risk curve, scatter diagram and heatmap in the validation set also showed the same results as the train set (Figure 4F-H). Then, we performed univariate and multivariate Cox regression analysis on the validation set, and the results showed that the risk score can still be used as an independent prognostic factor for patients with LUAD (Figure 5D, E). The AUC values of 1-, 3- and 5-year prognosis also proved good predictive ability (Figure 5F). And the AUC value of the total sample also proves this result again (Figure 5G).
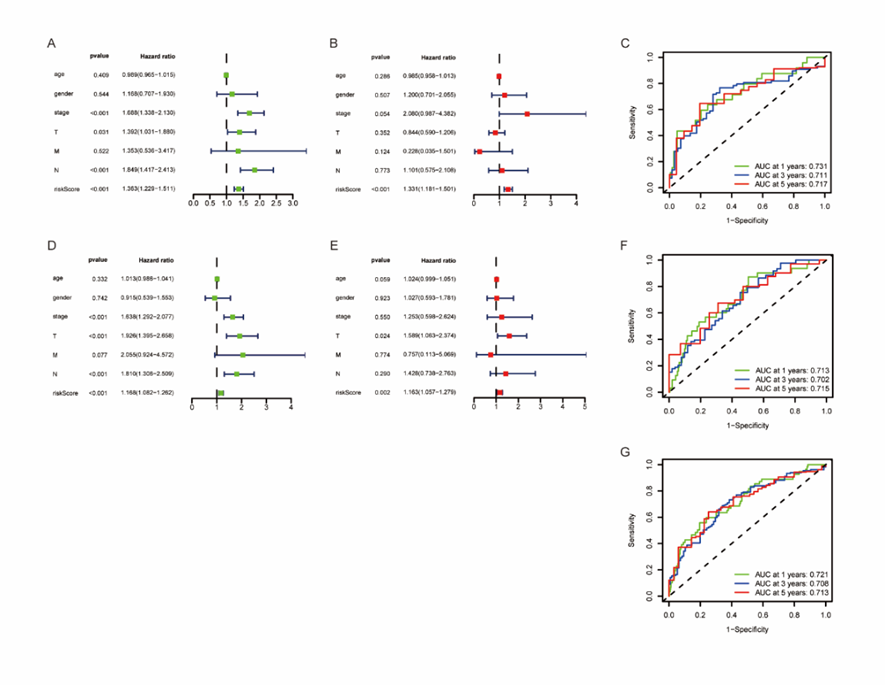
Figure 5: ROC and Cox analysis of risk characteristics based on 10 Characteristic Genes.
(A, B) Univariate Cox regression analysis and multivariate Cox regression analysis in the train set demonstrated that Risk Score was an independent risk factor for OS.
(C) ROC analysis of overall survival for the 10-gene signature in train cohort.
(D, E) Univariate Cox regression analysis and multivariate Cox regression analysis in the train set.
(F) ROC analysis of overall survival in the test set.
(G) Total ROC curve of train set and test set samples.
Correlation evaluation between clinical characteristics and survival model
We also grouped and tested the clinical features, the survival analysis after grouping still showed the same results as previously (Figure 6A). We can also see from the clinical correlation box graph that the risk scores of cluster1 and cluster2 and patients with different tumor stages also have significant differences (Figure 6B). In addition, according to the correlation heatmap, the expression level of apoptosis-related genes was significantly correlated with the risk score and two clusters, but not with the clinical characteristics (Figure 6C). Then, we studied the correlation between risk score and immunity, as shown in Figure 6D.

Figure 6: Correlation verification between risk model and clinical characteristics.
We can see from the figure, consistent with the foregoing, the infiltration of Dendritic cells and Mast cells decreased with the increase of risk score. In addition, Plasma cells, Monocytes and T cells CD4 memory showed the same trend. On the contrary, when the risk score increased, Macrophages M0, Macrophages M1, Neutrophils and NK cells showed an increasing trend. Thus, it can be seen that when the risk score increased, the body immunity showed a state of inhibition, indicating a poor survival prognosis. This result further proves the prediction ability of our model.
Lung cancer is a malignant tumor with highest incidence rate and mortality rate. However, the early diagnosis and treatment results are still not ideal, and the recurrence and metastasis rate after treatment are still not low. Thus, it is very important to study the pathogenesis of non-small cell lung cancer and find new method of diagnosis and treatment, especially LUAD, which accounts for a high proportion[12]. At present, the treatment of LUAD tends to be accurate and individualized[13, 14]. Nevertheless, in terms of the current clinical situation and treatment guidelines, the realization of accurate individualized treatment is very difficult. The surgical treatment of early LUAD patients did not achieve very ideal results. Lately, due to the widespread use of sequencing technology in cancer research, the diagnosis and treatment at the molecular level has gradually become a hot topic, and various prognostic predictors of LUAD have been proposed[15-18]. Using biomarkers to diagnose and predict survival prognosis has become a trend[13]. As a biological process, apoptosis is involved in the occurrence and development of tumors and plays an important role. As far as we know, there is still a lack of research on predicting the prognosis of LUAD patients with apoptosis-related genes. Therefore, it is of clinical significance to find apoptosis-related genes and establish prognosis prediction model.
In our study, we selected 10 apoptosis-related gene signatures based on enrichment analysis in MSigDB. Based on the expression level of these genes, we defined the apoptosis state of LUAD tissue and divided these tumor samples into two subgroups. Compared with cluster1, cluster2 showed apoptosis inhibition, immune cells infiltration also showed immune system inhibition, and the survival prognosis was significantly worse than cluster1. Then, based on TCGA database, a prognostic risk model of apoptosis-related genes in LUAD was established by using univariate and Lasso Cox regression analysis. Using this model, we can divide LUAD patients into high-risk and low-risk groups. Furthermore, by testing clinical data and risk model, we proved that risk score can be used as an independent predictor of LUAD. Univariate, multivariate Cox analysis and ROC curve results also proved this conclusion. We then demonstrated once again the predictive power of our prognostic model in the test set. Finally, we also analyzed the correlation between clinical characteristics and prognostic model. Our research may help clinicians make individualized and effective treatment decisions, and provide help for further research.
However, it should be acknowledged that there are still some deficiencies in our study. First, we established a prognostic model through traditional statistical methods and then evaluate it. Although these methods have been confirmed in some studies, there is still room for improvement in further research in the future. Second, in our study, further experiments are needed to clarify the biological function of apoptosis-related genes in LUAD, which may help us to deeply understand the specific mechanism.
In short, we established a new apoptosis-related survival model to stratify the risk of LUAD patients and evaluate their prognosis. The model is composed of 10 apoptosis-related genes (DNM1L, SOD1, TGFB2, BIRC3, KRT18, BIK, DNAJA1, TIMP1, BCL2L10, CYLD) in LUAD, and may help to realize the individualized treatment and management of LUAD patients.
We used R software (version 4.0.3) to perform all statistical analysis. And the statistical analysis in this study is mostly based on online database. The Kaplan-Meier survival analysis was used to evaluate the prognosis, Lasso-cox regression analysis to establish the model and Univariate and multivariate Cox regression analysis to verify whether it can be used as an independent factor. ROC curves were used to assess the predictive accuracy for survival. We considered P < 0>
Ethics approval and consent to participate
All the gene expression data used the data that had been normalized. For this study, only open-access data were used, which excluded the requirement of authorization from the Ethics Committee.
Release of consent
Not applicable.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors report no conflicts of interest in this work.
Funding
This study was funded by Natural Science Foundation of Zhejiang Province(LY21H160059)
Authors' contributions
Wei Zhang and wrote the manuscript. Wentao Zhu and Suwei Xu collected and analyzed the raw data. Wei Zhang and Yu Liu designed the whole work.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
