
Case report | DOI: https://doi.org/10.31579/2578-8868/328
1Department of Neurology and Clinical Neurophysiology, Base Hospital of the Federal District, Brasília, Distrito Federal, Brazil.
2Faculty of Medicine, Federal University of Piaui, Teresina, Piaui, Brazil.
3Department of Hematology, Base Hospital of the Federal District, Brasília, Distrito Federal, Brazil.
4Pathological Analysis Laboratory, Sabin Medicina Diagnóstica.
*Corresponding Author: Deborah Castro Ferreira de Oliveira, Department of Neurology and Clinical Neurophysiology, Instituto Hospital de Base do Distrito Federal, SMHS - Área Especial Q. 101 - Asa Sul, Brasília - DF, 70330-150, Brazil.
Citation: De Oliveira DCF, Ferreira De Oliveira IDC, De Carvalho MR, Eneas Bomtempo CS, Góes Louly Bustamante APD, et al, (2024), Erdheim-Chester Disease: Case Report with Brainstem Involvement and Mucocutaneous Manifestations, J. Neuroscience and Neurological Surgery, 15(5); DOI:10.31579/2578-8868/328
Copyright: © 2024, Deborah Castro Ferreira de Oliveira. This is an open-access article distributed under the terms of The Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Received: 21 June 2024 | Accepted: 01 July 2024 | Published: 11 July 2024
Keywords: erdheim-chester disease; histiocytosis, non-langerhans-cell; central nervous system; hematologic diseases; case reports
Erdheim-Chester Disease (ECD) is a malignant disease of myeloid progenitor cells that predominantly affects elderly men. ECD is considered a clonal disorder and inflammation corroborates to disease progression and organ injury. Clinical manifestations differ according to the extent and distribution of the involved sites. At diagnosis, most patients exhibit bone involvement, and the majority also present with involvement of at least one extraosseous site. This report describes a 54-year-old man who was diagnosed with ECD following histopathological examination of the sphenoid sinus mucosa.
Erdheim-Chester disease (ECD) is a malignant disease of myeloid progenitor cells, first described in 1930 as lipoid granulomatosis. [1,2,3] It is characterized by infiltration histiocytes into tissues, often due to mutations in the mitogen-activated protein kinase (MAP) pathways. ECD affects several organ systems, including the central nervous system and cardiovascular system and the bones, lungs, and kidneys. [1,2,4,5] ECD is three times more prevalent in adult men than women and typically presents between the sixth and seventh decades of life. [1]
Cells classified as members of the mononuclear system are macrophages, monocytes, dendritic cells. Histiocyte refers to tissue-resident macrophages.[6] Thus, histiocytosis is the term that defines the accumulation of cells derived from macrophage or dendritic cells. The most recent classification system of histiocytoses and neoplasms of the macrophage-dendritic cell lineages divide these diseases in five groups: Langerhans-related, cutaneous and mucocutaneous, malignant histiocytosis, Rosai–Dorfman disease, and hemophagocytic lymphohistiocytosis and macrophage activation syndrome. [6,7]
ECD was previously classified as non-Langerhans disease. However, the finding that nearly 20% of ECD have Langerhans cells lesions and, in both conditions, clonal mutations in MAPK genes pathway are seen made ECD be classified in the Langerhans-related group. [6]
ECD was initially considered an inflammatory condition. Now it is considered a clonal disorder and inflammation corroborates to disease progression and organ injury. [8] BRAF mutation was encountered in half of ECD biopsies with lesions showing the expression of the mutated protein in histiocytes. This mutation leads to gain-of-function and independent activation of the MAPK signalling pathway, cell proliferation, survival, and angiogenesis.[8] On the other hand, tissue samples from ECD patients evidence expression of cytokines and chemokines.[8]
In a recent literature review on neurological involvement, 50% of patients presented parenchymal lesions involving pituitary gland or hypothalamus; 35% had brainstem lesions, 25% had cerebellar involvement, 17.5% had dural involvement, and 10% had dural and parenchymal lesions discovered on MRIs. [9] CNS involvement carries a higher rate of morbidity and mortality. Common diagnostic modalities involve imaging, including brain MRI, tissue biopsies, and immunohistochemistry. Surgical cytoreduction is often necessary along with pharmacological treatment. [10] In this paper, we report a case of ECD diagnosed at our Neurological Center.
A 52-year-old Caucasian man presented with a one-year history of headache, decreased bilateral visual acuity, and right hemifacial paresthesia. Eight months prior to his presentation, an outside physician diagnosed him with trigeminal neuralgia and prescribed carbamazepine. Five months prior to his presentation, the patient had painless, yellowish plaques with defined contours on both eyelids (Figure 1).
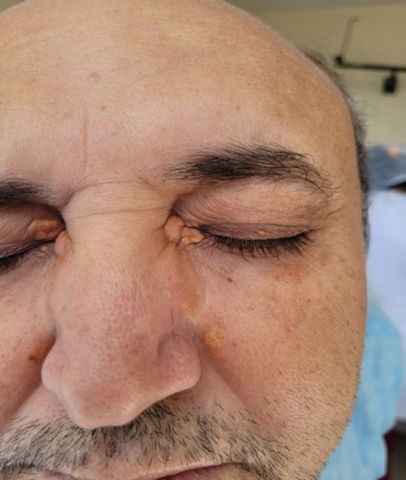
Figure 1: Xanthelasma, plaques in the bilateral eyelid region, with a well-defined contour, yellowish color, painless. Image authorized by the patient.
Upon admission to the neurology ward, the patient reported pulsatile headaches, hypogeusia, hyporexia, afternoon fever (unmeasured), and an 8-kilogram weight loss over the previous two months. Upon admission, the physical examination revealed bipalpebral lesions, right eye abduction deviation, a visual deficit in the left inferior medial field, left hypoacusis, exhaustible horizontal nystagmus in the left eye, and gait ataxia during the Tandem maneuver. Brain magnetic resonance imaging (MRI) revealed multiple focal hyperintense lesions on T2/FLAIR images (Figure 1) with adjacent edema affecting the entire brain stem and chronic inflammatory lesions in the maxillary, ethmoid, and sphenoid sinuses. Laboratory tests, including a complete blood count, inflammatory markers, and electrolyte levels, were normal. Serologies for human immunodeficiency virus, syphilis, hepatitis B, and hepatitis C were non-reactive. A cerebrospinal fluid analysis revealed no significant abnormalities. Radiographs of the long bones revealed sclerotic lesions compromising the bone marrow and cortical thickening with a symmetrical appearance in the heads of the humeri (Figure 2), heads and necks of the femurs, distal femurs, and tibias from the plateaus to the distal diaphyses (Figure 2).
Contrast-enhanced computed tomography (CT) of the chest revealed material with the density of soft tissue involving the aortic branches, consistent with ECD. A discrete pulmonary interstitial component and pericardial effusion were also noted. Abdominal CT with contrast demonstrated an extensive retroperitoneal lesion encompassing the kidneys, adrenal glands, aorta, and vena cava (Figure 2), which are also compatible with ECD.
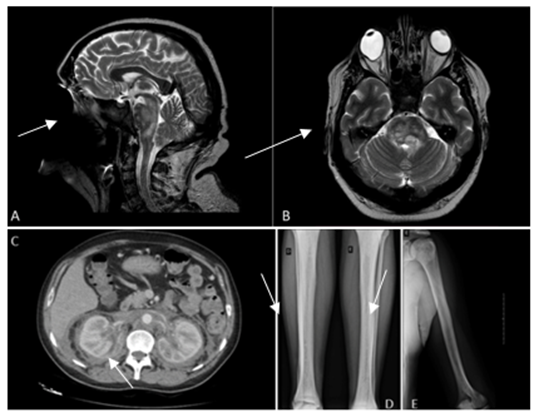
Figure 2: A) A contrast-enhanced magnetic resonance image (MRI) of the head obtained prior to presentation is shown (sagittal, T2-weighted). Arrow: multiple hyperintense focal lesions affecting the brainstem. B) A contrast-enhanced head MRI obtained prior to presentation is shown (axial, T2-weighted). Arrow: multiple hyperintense focal lesions affecting the brainstem. C) A contrast-enhanced abdominal computed tomography image is shown. Arrow: extensive retroperitoneal lesion. D) Radiography of the lower limbs is shown. Arrow: sclerotic lesions compromising the bone marrow symmetrical-looking cortical thickening. E) Radiography of the left arm is shown. Arrow: sclerotic lesions compromising the bone marrow symmetrical-looking cortical thickening.
The anatomopathological examination of the sphenoid sinus mucosa (Figure 3) revealed fibrosis and a chronic inflammatory infiltrate including lymphocytes, plasma cells, histiocytes with foamy cytoplasm, and neutrophils. There was also hyperplasia of the seromucinous glands. Tests for acid-fast bacilli and yeast were negative. immunohistochemical analysis revealed histiocytes with cytoplasmic CD68 staining and no S-100 staining, suggesting histiocytic proliferation and supporting the clinical diagnosis of ECD. A BRAF mutation was detected. Subsequently, the patient was transferred to the hematology department and administered cladribine (0.12 mg/kg for 4– 6 cycles).
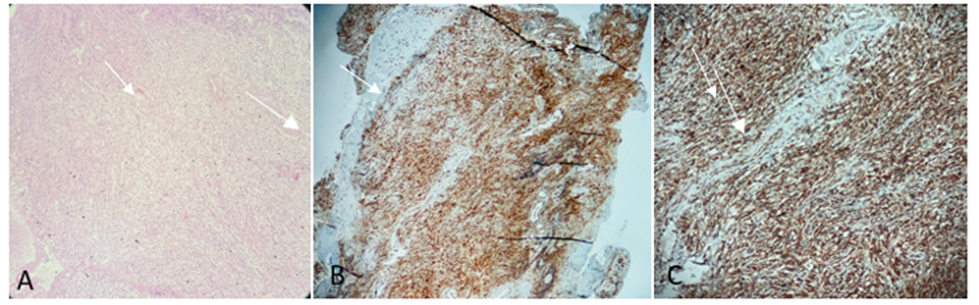
Figure 3: A) Hematoxylin and eosin analysis shows lymphocytic chronic inflammatory infiltrate. B) Ulcerated sphenoid sinus mucosa, with mixed inflammatory infiltrate, associated with dense histiocytic infiltrate based on CD-68 positive and S-100 negative histiocytes, is shown. C) Cytoplasmic staining for CD-68 is shown.
ECD is a multisystemic clonal disorder of the myeloid lineage characterized by the proliferation of foamy histiocytes and Touton giant cells in various tissues and organs. These cells produce pro-inflammatory cytokines and chemokines and harbor somatic mutations such as BRAF V600E. [1,5]
This case report illustrates the occurrence of Erdheim–Chester disease in a brazilian patient presenting with multisystem and neurological manifestations. Our patient presented sociodemographic characteristics similar to findings in other case series.[7]
At diagnosis, most patients exhibit bone involvement, and the majority also present with at least one extra-osseous site of involvement.[1] While some patients are asymptomatic, others experience a rapidly progressive clinical course. Bone manifestations are the most common clinical features, followed by kidney involvement, periaortic encasement, hypogonadism, lung involvement, diabetes insipidus, and central nervous system infiltration.[11] Symmetrical diaphyseal and metaphyseal osteosclerosis of the lower extremities is almost always present. Radiographically, the disease typically presents as bilateral and symmetrical osteosclerosis of the long bones. [1,9]
Neurological involvement, either intra- or extra-axial, is observed in approximately half of patients with ECD. [1,12] Central nervous system involvement serves as an independent predictor of a worse outcome[13] Unilateral or bilateral infiltration of the orbits, presenting as exophthalmos, retro-orbital pain, oculomotor paralysis, or blindness, occurs in approximately 25% of patients. [14] Approximately 50% of patients with ECD have parenchymal lesions affecting the cerebral hemispheres, pituitary gland, or hypothalamus; 35% have brainstem lesions; 25% have cerebellar involvement; 17.5% have dural involvement; and 10% have both dural and parenchymal lesions on MRI. [15] A recent case series addressing cerebellar manifestations showed that involvement of the cerebellar hemispheres and peduncles, pons, midbrain and cerebral peduncles was common in all patients; neurophysiological studies showed damage to sensory and motor pathways; cognitive changes at admission and during follow-up. [7]
The diagnostic evaluation for ECD must include an anatomopathological study in addition to laboratory and imaging tests. Foamy histiocytes laden with lipids and exhibiting a distinct immunophenotype within an appropriate cellular and/or fibrotic matrix are typically observed on anatomopathological examination. [6] Biopsy samples should undergo molecular testing using a genetic panel or next-generation sequencing to identify BRAF V600E and other mutations, including NRAS, KRAS, ARAF, PIK3CA, MAP2K1, and ALK. The immunohistochemical analysis is typically positive for CD14 (a lipopolysaccharide receptor), CD68 (a lysosomal macrosialin), CD163 (a hemoglobin and haptoglobin scavenging receptor), Factor XIIIa (tissue glutaminase), and fascin (actin-binding protein). [13]
Treatment is necessary in symptomatic patients or in those with evidence of central nervous system involvement or organic dysfunction. [6] Upon detection of a mutation, targeted treatment with vemurafenib, a BRAF inhibitor, or cobimetinib, a MEK inhibitor, may be initiated. Cladribine is the preferred cytotoxic chemotherapy. [13,15]
ECD is rare and has a diverse presentation, posing diagnostic challenges that often result in late or erroneous diagnoses. The diagnosis of ECD relies on the identification of distinct histopathological findings within an appropriate clinical and radiological context. Increased awareness of this disease may decrease the number of underdiagnosed and undertreated cases. In Brazil, data on this disease and details on its involvement and treatment are scarce. To our knowledge, this is the first report of ECD in central-western Brazil.
We would like to thank Editage (www.editage.com.br) for English language editing.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
