
Case Report | DOI: https://doi.org/10.31579/2692-9406/002
*Corresponding Author: Luís Teles, Internal Medicine Department, Centro Hospitalar de Entre o Douro e Vouga, R. Dr. Cândido Pinho nº54520-211 Santa Maria da Feira, Portugal.
Citation: Luís T, Bernardo M, António M. (2020) Erdheim-Chester Disease: Case Report with Aggressive Multiple Organ Manifestations. Biomedical Research and Clinical Reviews. 1(1); DOI: 10.31579/2692-9406/002
Copyright: ©2020 Luís Teles, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 07 February 2020 | Accepted: 25 June 2020 | Published: 30 June 2020
Keywords: non-langerhans cell; multiple organ; braf v600e
Erdheim-Chester disease (ECD) is a rare non-Langerhans cell, lipid-laden histiocytosis with specific histological and radiological findings. The diagnosis sometimes is established lately in the course of the disease.
We present a case of a 64-year-old female with elevated inflammatory markers for one year and symptoms related with her comorbidities, particularly bone pain and short of breath. Past medical history includes a stage III chronic kidney disease, central diabetes diagnosed when she was 38 years old, Paget Disease, metabolic syndrome and ischemic cardiopathy. Computed tomography in the near past showed a tissue densification in the thoracic vertebral column and kidneys with hairy aspect. X-ray of the arms, legs, skullcap, and demonstrated sclerotic changes. F-fluorodeoxyglucose positron emission tomography showed uptake in the skull, mediastinum, abdomen and long bones from arms and legs. Biopsy of the hairy kidney was consent after 4 years of an unknown disease in progression. Histological findings of the biopsy reported a diffuse infiltration by foamy histiocytes. On immunohistochemical staining, the histiocytes were positive for CD68 and negative for CD1 and S100. Mutation of BRAF V600E was present and ECD was established. Tocilizumab was initiated off label due to psychiatric contra indication for interferon use and no clinical conditions for BRAF inhibitors and symptoms started being controlled.
The diagnosis of ECD is usually challenging due to the rarity of the disease and clinical overlapping with many other conditions. The rarity and variable presentation of this disease usually leads to delayed diagnosis and to high morbidity and mortality rates from associated complications.
Introduction
Erdheim-Chester disease (ECD) is a rare non-Langerhans cell, lipid-laden histiocytosis with specific histological and radiological findings. [1] Its estimated there are 600 cases described in the literature. The disease usually presents between 40 and 60 years and its etiology is still unknown but BRAF proto-oncogene has been identified in the majority of ECD cases.
The diagnosis of ECD is made with a combination of clinical presentations and imaging features, that may be present on other diseases, that is why the diagnose sometimes is established lately in the course of the disease. Heterogenous clinical manifestations depend on site and degree of involvement, but around 95% of ECD patients have skeletal involvement and bone pain is the main symptom. Apart from skeletal involvement, ECD may infiltrate eyes, endocrine organs, kidney, heart and central nervous system (CNS). [2]
The differential diagnosis of ECD includes diseases such as Langerhans cell hystiocytosis, multiple sclerosis, Paget disease, amyloidosis and others.
The first line treatment is interferon alfa although it might not be eligible for all the patients depending of the comorbidities and efficacy is limited against severe manifestations, specifically CNS and cardiovascular involvement. Treatment with other agents is based on case reports described in the literature.
In this paper, we present an ECD case with multiple organs involved because of the delay between the initial symptoms and the diagnosis. To the best of our knowledge, this is one of the few cases reported from Portugal.
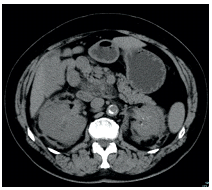
A 64-year-old female presented with elevated inflammatory markers for one year and symptoms related with her comorbidities, particularly bone pain and short of breath. Past medical history includes a stage III chronic kidney disease, diabetes insipidus diagnosed when she was 38 years old, Paget Disease, metabolic syndrome and ischemic cardiopathy.On admission to our appointment, the patient’s general appearance was normal and hemodynamically stable. The initial laboratory workup revealed mild anemia (11.2 g/dL), normal white blood cell count and normal platelets. Chronic kidney disease was supported with creatinine of 1.6 mg/dL. Liver function was normal. The inflammatory markers such as C-reactive protein (CRP) and the erythrocyte sedimentation rate (ESR) were elevated - CRP: 106 mg/dL (reference values <5.0 mg/dL); ESR 93 mm (reference values <20 mm). Patient had made a computed tomography (CT) in the near past that showed an anterior tissue densification in the thoracic vertebral column and kidneys with hairy aspect (Figure 1).
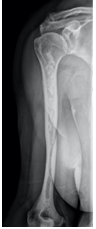
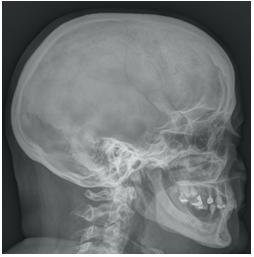
The patient underwent radiological studies, with bilateral (Figures 2 and 3).
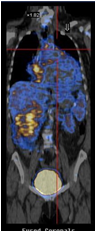
F-fluorodeoxyglucose positron emission tomography (FDG-PET) showed uptake in the skull, mediastinum, abdomen and long bones from arms and legs (Figure 4).
Cardiac MRI revealed a fibrotic structure next to the free wall of the right atrium.After these results we started questioning the past medical history of Paget Disease and diabetes insipidus and thought these findings probably would come from the same disease.A kidney and a bone biopsy were proposed but patient denied it for 4 years until she was admitted to the ward because of a cardiac dysthymia (needing definitive pacemaker) with decompensated heart failure, aggravated pericardium effusion and acute on chronic kidney failure. Surgery was proposed for intraperitonealization of the ureters and biopsy of the hairy kidney was consent after 4 years of an unknown disease in progression.Histological findings of the biopsy reported a diffuse infiltration by foamy histiocytes. On immunohistochemical (ICH) staining, the histiocytes were positive for CD68 and negative for CD1 and S100. Mutation of BRAF V600E was present and ECD was established.Once the diagnose of ECD was confirmed and because of its agressive nature and involvement in this patient treatment was started immediately. She had a psychiatric contra indication for interferon use and had no clinical conditions for BRAF inhibitors. She started treatment with tocilizumab 8mg/kg/month intravenous based on its use described in case reports.Our patient had a good response after starting tocilizumab. Until now she is under surveillance with a stable disease.
ECD is a rare, multi system histiocytic neoplasm characterized by tissue infiltration or organ encasement by CD68+, CD1-, S100-low/negative foamy histiocites. Heterogenous clinical manifestations depend on site and degree of involvement. Skeletal involvement with sclerotic lesions and radiotracer uptake is common. Other disease manifestations include neurologic symptoms due to CNS involvement, diabetes insipidus, constitutional symptoms and heart and lung involvement. When affected, CNS, lungs, heart and retroperitoneal space account for a severe prognosis. [3]
In our patient diabetes insipidus was diagnosed when she was 38 and it might be the first manifestation of ECD, misinterpreted for almost 30 years. The diagnosis of ECD is usually challenging due to the rarity of the disease and clinical overlapping with many other conditions. In our case patient was being followed in orthopedic appointment because of bone lesions thought to be related with a Paget Disease. The rarity and variable presentation of this disease usually leads to delayed diagnosis and to high morbidity and mortality rates from associated complications.
With the progression of the disease until the diagnose is established sometimes patients may not be eligible for first line treatments, as occurred with our patient where we used tocilizumab based on case reports.
The authors have no funding or conflicts of interest to disclose.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
