
Research Article | DOI: https://doi.org/10.31579/2690-1919/459
1 Department of Internal Medicine, National Taiwan University Hospital, Hsin-Chu Branch, Taiwan.
2 Department of Food Science, National Chiayi University, Chiayi, Taiwan.
3 Department of Biochemical Science and Technology, National Chiayi University, Chiayi, Taiwan.
# Qiao-Zhen Chang, and Bo-Cheng Mu contributed equally.
*Corresponding Author: Yun-Wei Lin, Consultant Division of pediatric orthopedics, Department of Biochemical Science and Technology, National Chiayi University, Chiayi, Taiwan.
Citation: Jen-Chung Koa, Jyh-Cheng Chenb, Ching-Hsiu Huangb, Pei-Jung Chenc, Yun-Wei Linc, et al., (2025), Epigallocatechin-3-Gallate Potentiates the Cytotoxic Effect of Erlotinib in Non-Small-Cell Lung Cancer Cells by P38 Mapk Mediated Xrcc3 Downregulation, J Clinical Research and Reports, 18(1); DOI:10.31579/2690-1919/459
Copyright: © 2025, Yun-Wei Lin. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 11 December 2024 | Accepted: 06 January 2025 | Published: 16 January 2025
Keywords: epigallocatechin gallate; xrcc3; erlotinib; cytotoxicity; non-small-cell lung cancer
Green tea polyphenols, particularly (-)-epigallocatechin gallate (EGCG), are thought to be responsible for the major physiologic benefits. XRCC3 participates in homologous recombination (HR) to repair DNA double-strand breaks (DSBs). The use of EGCG in combination with erlotinib to increase survival in patients with non-small-cell lung cancer (NSCLC) is still debatable and requires further research.
Materials and Methods
We used the MTS, trypan blue dye exclusion and colony-formation ability assay to determine whether EGCG alone or in combination with erlotinib had cytotoxic effects on the four NSCLC cell lines. Real-time PCR was conducted to measure the amounts of XRCC3 mRNA. The protein levels of phosphorylated p38 MAPK and XRCC3 were determined by Western blot analysis.
Results
We demonstrated that EGCG can amplify erlotinib cytotoxicity in NSCLC cells. EGCG treatment promote p38 MAPK activation and decreased XRCC3 mRNA and protein levels; moreover, combined treatment with erlotinib resulted in a significant reduction in XRCC3 protein levels and triggered XRCC3 degradation via a 26S proteasome-dependent pathway. The p38 MAPK activity was greatly increased by constitutively active MKK6 vector expression (MKK6E) and could promote XRCC3 degradation, which enhanced erlotinib and EGCG cytotoxicity. Conversely, SB203580, a p38 MAPK inhibitor, could prevent p38 MAPK activation and restore XRCC3 levels that had been decreased by erlotinib and improve cell survival.
Conclusion
Overall, our findings imply that XRCC3 downregulation is one of the primary mechanisms boosting the lethal effects of erlotinib by EGCG in NSCLC cells.
When single-stranded DNA (ssDNA) invades a homologous duplex DNA, which is fueled by the RAD51/RecA family of ATPases, homologous recombination (HR) pathways, are involved in the repair of DNA double-strand breaks (DSBs). Rad51 is a key component of HR pathways; ssDNA and RAD51 combine to create a nucleoprotein filament that performs homology search and catalyzes strand exchange [1]. To induce stable filament assembly and/or nucleate Rad51 filament formation on ssDNA in vivo, numerous variables are needed. RAD51 paralogs, which only slightly resemble RAD51 in the ATPase domain, are one of the elements contributing to this process. RAD51B [2], RAD51C [3], RAD51D [4], or complementation of radiation-sensitive Chinese hamster cell mutants XRCC2 [5] and XRCC3 [6] are the five canonical Rad51 paralogs in mammalian cells. The paralogs have been demonstrated to form two complexes biochemically, i.e., RAD51C–XRCC3 and RAD51B–RAD51C–RAD51D–XRCC2 [7]. At DSB sites, XRCC3 encourages Rad51 assembly [8]. To create a D-loop, the resulting RAD51-ssDNA filaments invade other unharmed homologous sequences. Eventually, gene conversion is the outcome of DNA synthesis from the invasive strand [9].
Non-small-cell lung cancer (NSCLC), which makes up roughly 85% of all lung cancer cases, has the highest mortality rate worldwide [10]. A shorter survival and poor prognosis are linked to tyrosine kinases (TKs) of the epidermal growth factor receptor (EGFR) or their ligands in NSCLC [11]. To prevent EGFR activation, small compounds known as TK inhibitors, such as erlotinib, can block the ATP-binding site on the receptor’s catalytic domain [12].
Green tea is made from the leaves of Camellia sinensis, a member of the Theaceae family, and exerts positive effects on human health, including anticancer, anti-obesity, antidiabetic, anticardiovascular, anti-infectious, and anti-neurodegenerative properties [13]. The majority of these biological effects are thought to be caused by (-)-epigallocatechin gallate (EGCG), the most prevalent catechin in green tea and the subject of earlier research. Moreover, 240–320 mg of catechins, of which 60%–65% is made up of EGCG, are present in a cup of green tea that is generally brewed using 2.5 g of tea leaves [14]. In the study by Wang et al., the extent of colon cancers in rats receiving dimethylhydrazine was reduced by EGCG [15]. EGCG, a well-known antioxidant, inhibits reactive oxygen species (ROS), which encourages oxidative DNA damage, mutagenesis, and tumor development and has anticancer effects [16], because EGCG-induced ROS production can activate AMPK by upregulating Ca2+/calmodulin-dependent protein kinase (CaMKK) and/or liver kinase B1 (LKB1) [17, 18], which inhibits the mechanistic target of rapamycin kinase (mTOR), having anticancer effects.
EGCG lessens side effects and strengthens anticancer effects; thus, it has been shown to have an additive or synergistic effect with chemopreventive medicines [19]. In previous study, EGCG inhibits cell proliferation and migration in NSCLC harboring wild-type EGFR (A549 cell line), in-frame deletion of exon 19 mutation (HCC827 cell line), and L858R/T790M double mutation (H1975 cell line) [20]. Moreover, the co-administration of EGCG and EGFR TK inhibitor gefitinib synergistic suppresses the tumor growth in the A549 xenograft mouse model [21]. In this study, we describe how EGCG affects XRCC3 expression and EGCG cytotoxicity in NSCLC cells. We demonstrate that erlotinib and EGCG combined therapy can lower XRCC3 protein levels in lung cancer cells by activating p38 MAPK. EGCG cytotoxicity is enhanced when the p38 MAPK activity is amplified and XRCC3 expression is downregulated. By modifying the XRCC3 and p38 MAPK signaling pathways, this study may offer a novel and more effective therapeutic approach to lung cancer treatment.
Chemicals and antibodies
EGCG was obtained from Sigma Chemical (St. Louis, MO, USA). MG132, SB203580, and N-acetyl-Leu-Leu-norleucinal (ALLN) were provided by Calbiochem-Novabiochem (San Diego, CA, USA). Phospho-p38 MAPK (Thr180/Tyr182) and phospho-MKK3 (Ser189)/MKK6 (Ser207) antibodies were purchased from Cell Signaling (Beverly, MA, USA). Abcam (Santa Cruz, CA) provided the rabbit polyclonal antibodies against XRCC3 (ab103070). Santa Cruz Biotechnology (TX, USA) provided the antibodies against p38 (C-20) (sc-535), MKK3 (N-20) (sc-959), and Actin (I-19) (sc-1616). Abcam provided the antibodies against XRCC3 (ab103070).
Cell lines and culture
RPMI-1640 complete medium supplemented with sodium bicarbonate (2.2% w/v), l-glutamine (0.03% w/v), penicillin (100 units/mL), streptomycin (100 µg/mL), and fetal calf serum (10%) was used to culture the human lung carcinoma cells H520, H1703, A549, and H1975, which were obtained from the American Type Culture Collection (Manassas, VA, USA).
Western blot analysis
After different treatments, the cells were rinsed twice with cold phosphate-buffered saline (PBS) solution and lysed in whole-cell extract buffer (20 mM HEPES [pH 7.6]), 75 mM NaCl, 2.5 mM MgCl2, 0.1 mM EDTA, 0.1% Triton X-100, 0.1 mM Na3VO4, 50 mM NaF, 1 µg/mL leupeptin, 1 µg/mL aprotinin, 1 µg/mL pepstatin, and 1 mM 4-(2- Equal amounts of proteins from each set of experiments were subjected to Western blot analysis as previously described [22].
Transfection with small-interfering RNA and MKK6E vectors
Sense-strand sequences for XRCC3 (s14948) siRNA duplexes were purchased from Thermo Fisher Scientific. By using Lipofectamine 2000 (Invitrogen), siRNA duplexes (200 nM) were transfected into cells for 24 h. The transfection of plasmids was accomplished as previously mentioned. Exponentially expanding human lung cancer cells (106) were plated for 18 h before Lipofectamine was used to transfect NSCLC cells with MKK6E (a constitutively active version of MKK6), as previously described [23].
Quantitative real-time polymerase chain reaction (PCR)
PCR tests were carried out on an ABI Prism 7900HT, as directed by the manufacturer. The SYBR Green PCR Master Mix (Applied Biosystems, MA, USA) was used to detect the amplification of particular PCR products. Data were standardized to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (gapdh) for each sample. XRCC3 forward primer, 5'- AACCCGCGGGAGAGTCCCCA -3'; XRCC3 reverse primer, 5'- AAAGCCTGTGGGAGGCCCGA -3'; gapdh forward primer, 5'- CATGAGAAGTATGACAACAGCCT -3'; and gapdh reverse primer, 5'- AGTCCTTCCACGATACCAAAGT -3' were used in this study. Analysis was performed using the comparative CT value approach. The results were standardized to the housekeeping gene Gapdh for each sample.
Cell viability assay
An in vitro 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenol)-2- (4-sulfophenyl) Tetrazolium (MTS) assay was conducted. On 96-well tissue culture plates, 5000 cells were cultivated for each well. Drugs were introduced after plating to measure cell viability. After the culture period, 20 µL of the MTS solution (CellTiter 96 Aqueous One Solution Cell Proliferation Assay; Promega, Madison, WI, USA) was added. The cells were then cultured for another 2 h, and the absorbance was measured at 490 nm using an ELISA plate reader (Biorad Technologies, Hercules, CA, USA).
Combination index analysis of drug interactions
The effects of a 1:1 or 2:1 mixture of EGCG and erlotinib were examined using the MTS assay. To evaluate if a combination is synergistic (combination index [CI] < 0 xss=removed> 1.1), the CI, which considers the complete shape of the cell viability curve, was calculated using the computer program Calcusyn (Biosoft, Oxford, UK). The means of the CI values at fractions affected (FA) of 0.90, 0.75, and 0.50 were used to determine the difference between the three independent experiments.
Trypan blue dye exclusion assay
Erlotinib and/or EGCG were given to the cells for 24 h. Trypan blue dye cannot penetrate living cells, but it can enter dead ones. The number of trypan blue-stained cells and the percentage of dead cells were counted using a hemocytometer.
Colony-forming ability assay
Cells were trypsinized to count the number of cells right after the drug treatment and then washed with PBS. For each treatment, the cells were seeded in triplicate at a density of 500–1000 cells per 60 mm Petri dish. The cell colonies were stained with 1% crystal violet solution in 30% ethanol after the cells had grown for 10–14 days. Cytotoxicity was calculated by dividing the number of colonies in the treated cells by the number of colonies in the untreated control.
Experiments were conducted three or four times for each protocol. The results are expressed as the mean and the standard error of the mean (SEM). SigmaPlot 14.0 was used (Systat Software, San Jose, CA, USA). IBM SPSS Statistics for Windows version 20.0 (IBM Corp., Armonk, NY, USA) was used to examine the data. To see how treatments and time affected changes in cell viability, one-way analysis of variance was used to analyze differences between the two groups. This was followed by a Bonferroni post hoc test to ensure that all variables had a normal distribution. Differences were deemed significant at P < 0>
EGCG and erlotinib inhibited cell survival in NSCLC cells
We used the MTS assay to first determine whether EGCG alone or in combination with erlotinib had cytotoxic effects on the four NSCLC cell lines H520 (wild-type EGFR), H1703 (wild-type EGFR), A549 (wild-type EGFR), and H1975 (L858R/T790M). NSCLC cells dose- and time-dependently responded to EGCG, as depicted in Figure. 1A. In addition, the combined therapy of erlotinib (0.5–10 mM) and EGCG (0.5–20 mM) promote lung cancer cell cytotoxicity than either drug used alone (Figure. 1B). The CI was determined to ascertain the type of additive effect from EGCG and erlotinib (Table 1).
| Drug combination | CI Values Calculated for Different Effects | ||||||
| Cell line | (erlotinib:EGCG) | ED50 | ED75 | ED90 | |||
| H520 | (1:1) | 0.70 | ±0.03 | 1.51 | ±0.08 | 3.27 | ±0.42 |
| (1:2) | 0.74 | ±0.03 | 1.76 | ±0.22 | 4.67 | ±1.23 | |
| H1703 | (1:1) | 0.65 | ±0.07 | 1.01 | ±0.16 | 1.59 | ±0.40 |
| (1:2) | 0.29 | ±0.02 | 0.30 | ±0.01 | 0.34 | ±0.01 | |
| A549 | (1:1) | 0.35 | ±0.03 | 0.37 | ±0.07 | 0.40 | ±0.12 |
| (1:2) | 0.63 | ±0.16 | 1.57 | ±0.46 | 3.93 | ±1.32 | |
| H1975 | (1:1) | 0.50 | ±0.06 | 0.96 | ±0.08 | 1.85 | ±0.20 |
| (1:2) | 0.14 | ±0.02 | 0.20 | ±0.02 | 0.28 | ±0.03 | |
Data represent meanCI calculated fromthree independent experiments ±standard deviations | |||||||
| CI < 0.9 indicates synergism, CI = 0.9-1.1indicates additivity, and CI > 1.1 antagonism. | |||||||
Table 1: Values of combination index (CI) for erlotinib-EGCG interactions at different effects: ED50, ED75 and ED90.
The interactions between EGCG and erlotinib were primarily additive and antagonistic at the effect levels represented as ED90 and ED75; however, at ED50, they proved to be synergistic (CI < 0.1). On the evaluation of NSCLC cell death using the trypan blue exclusion assay, EGCG and erlotinib combined therapy had an additive effect on cell death (Figure. 1C). Moreover, erlotinib and EGCG combined therapy affected cell survival for up to 3 days in NSCLC cell lines (Figure. 1D). In these four NSCLC cells, the EGCG and erlotinib combined therapy inhibited cell growth more than erlotinib alone (Figure. 1D). The colony-formation assay was then used to determine if erlotinib and/or EGCG affect longterm clonogenic cell survival. In H520 (22.57% vs. 74.00%), H1703 (45.73% vs. 73.53%), A549 (45.97% vs. 70.83%), and H1975 (30.23% vs. 63.13%) cells, the colony-forming ability of EGCG and erlotinib combined treatment was lower than that of erlotinib treatment (Figure. 2).

Figure 1: Figure 1: Treatment of EGCG alone or combination with erlotinib response to cell survival in NSCLC cells. (A) For 24, 48, and 72 h, cells were exposed to EGCG at different concentrations (2.5, 5, 10, 20, 40, 60, 80, and 100 µM). The MTS assay was used to measure cell viability. (B) EGCG enhanced erlotinib cytotoxicity in NSCLC cells. The MTS assay was used to determine the cell viability after EGCG and erlotinib combined therapy at a ratio of 1:1 (upper panel) or 1:2 (lower panel). Three separate trials were used to gather the results (mean ± SEM). Using the one-way ANOVA to compare cells treated with erlotinib alone vs. EGCG/erlotinib combination, [a] ** signifies p < 0.01. Using the one-way ANOVA to evaluate cells treated with EGCG alone against EGCG and erlotinib combination, [b] ** p <0.01. (C) Unattached and attached cells were collected and stained with trypan blue after cells had been exposed to various dosages of EGCG (10 µM)/erlotinib for 24 h, and dead cells were manually counted. The population of dead cells was indicated by the proportion of trypan blue-positive cells, and the standard error was computed from three separate experiments. Using one-way ANOVA, the difference between cells treated with erlotinib alone vs. the EGCG/erlotinib combination, **p <0.001, *p < 0.05. (D) The number of living cells was counted using the MTS assay after the cells had been exposed to EGCG (20 µM) and erlotinib (10 µM) for 24, 48, and 72 h. Three separate trials were used to generate the results (mean ± SEM). Comparing cells treated with EGCG, erlotinib, or their combination revealed **p <0.01, using Student’s t-test.
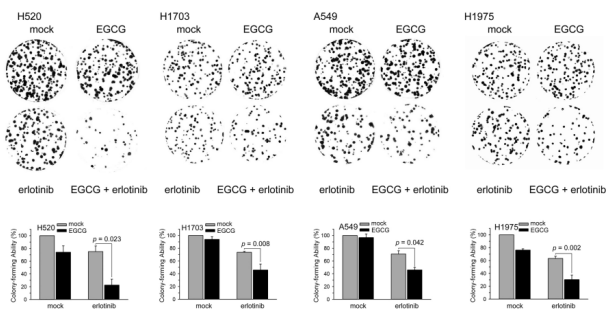
Figure 2: EGCG reduced erlotinib-induced colony-formation ability in NSCLC cells. After being exposed to EGCG (20 µM) and/or erlotinib (10 µM) for 24 h, cells were tested for their capacity to form colonies using the colony-formation assay. Three separate trials were used to gather the results (mean ± SEM). ** p < 0.01 when comparing cells treated with erlotinib alone against EGCG/erlotinib combination using a one-way ANOVA.
EGCG and erlotinib combined therapy additively decreased cellular XRCC3 protein levels
To examine the molecular mechanisms of EGCG in enhancing the cytotoxic effect of erlotinib on human lung cancer cell lines, NSCLC cells were exposed to various concentrations of EGCG alone or erlotinib and EGCG combination for 24 h. The protein levels of phosphorylated p38 MAPK and XRCC3 were then determined by Western blot analysis. EGCG dramatically decreased the levels of XRCC3 protein but increased the levels of phosphorylated p38 MAPK (Figure. 3A). In addition, EGCG and erlotinib combined treatment reduced XRCC3 levels in NSCLC cells (Figure. 3C).
EGCG decreased XRCC3 mRNA levels in erlotinib-treated NSCLC cells
EGCG and erlotinib combined treatment was given to NSCLC cells for 24 h to determine if the treatment-induced reduction in XRCC3 protein levels occurred at the transcriptional level. Real-time PCR was conducted to measure the amounts of XRCC3 mRNA in total RNA. XRCC3 mRNA levels reduced by EGCG was dose-dependent manner (Figure. 3B). In 4 different NSCLC cell lines, EGCG and erlotinib combined treatment had a more suppressive effect on XRCC3 mRNA levels (Figure. 3D). Next, we investigated potential mechanisms for posttranscriptional regulation of XRCC3 transcripts under EGCG and/or erlotinib treatment. In the presence of actinomycin D (an inhibitor of RNA synthesis), EGCG and erlotinib combined treatment showed less XRCC3 mRNA stability than the erlotinib treated alone in H520, H1703, and H1975, but not in A549 cells (Figure. 3E; Table 2).

Figure 3: In human NSCLC cell lines, erlotinib and/or EGCG decreased XRCC3 mRNA and protein levels. (A, B) NSCLC cells (106 ) were grown in full media for 18 h before being treated with various EGCG doses for 24 h. Following treatment, total RNA was extracted and submitted to real-time PCR for examination of XRCC3 mRNA expression and Western blot analysis for the quantification of the levels of XRCC3, phospho-MKK3/6, phospho-p38 MAPK, actin, MKK6, and p38 MAPK proteins. (C) Erlotinib (5 and 10 µM) and/or EGCG (20 µM) were administered to NSCLC cells (106 ) after they had been grown in full medium for 18 h. The treatment lasted for 24 h. Following treatment, cell extracts were utilized to evaluate the levels of XRCC3 protein after the aforementioned treatment. Four investigations are represented by Western blots. (D) Cell extracts were utilized to evaluate the amounts of XRCC3 mRNA after the aforementioned treatment. Total RNA was extracted, and real-time PCR was used to ascertain XRCC3 mRNA expression. Four separate experiments were conducted, and the findings (means standard deviation) were collected from each. Using the oneway ANOVA to compare cells treated with EGCG and erlotinib separately or together, ** p < 0.01. (E) NSCLC cells were treated with EGCG (20 µM) and/or erlotinib (10 µM) for 24 hours in the presence or absence of actinomycin D (1 µg/mL) for 3, 6, or 9 hours. The total RNA was isolated, and XRCC3 mRNA expression levels were determined using real-time PCR. The results (mean ± SEM) were acquired from three independent experiments. (F) Treatment with erlotinib plus EGCG causes XRCC3 to be proteolyzed by 26S proteasome. EGCG (20 µM) and erlotinib (10 µM) were administered to NSCLC cells for 18 h, followed by a 6-h co-treatment with the 26S proteasome inhibitors MG132 (10 µM) or ALLN (10 µM). For Western blotting, entire cell extracts were collected. Four investigations are represented by Western blots.
| XRCC3 mRNA half-life (h) | ||||
| cells | control | EGCG | erlotinib | EGCG+erlotinib |
| H520 | 7.89 | 6.19 | 6.98 | 5.34 |
| H1703 | 8.71 | 5.57 | 7.13 | 3.87 |
| A549 | 3.96 | 3.34 | 3.50 | 2.81 |
| H1975 | 8.71 | 4.99 | 7.04 | 3.92 |
Table 2: XRCC3 mRNA half-life (h) for EGCG and/or erlotinib treatment.
EGCG and erlotinib combined treatment decreased XRCC3 protein levels via 26S proteasome-directed proteolysis
To determine if the 26S proteasome was responsible for the degradation of the XRCC3 protein, MG132, or ALLN, two 26S proteasome inhibitors were co-added to NSCLC cells that had been exposed to erlotinib and EGCG. Consequently, MG132 and ALLN stopped the XRCC3 protein degradation induced by EGCG and erlotinib combined treatment (Figure. 3F). Together, EGCG and erlotinib combined treatment caused XRCC3 protein degradation through the 26S proteasome-dependent pathway, which was the mechanism for reducing XRCC3 protein levels.
Influence of the p38 MAPK signaling pathway on XRCC3 protein levels in lung cancer cells exposed to EGCG and erlotinib combined treatment
To evaluate the role of p38 MAPK activation in regulating XRCC3 expression, we investigated whether blocking p38 MAPK activity could
affect XRCC3 expression in NSCLC cells. NSCLC cells cultured with a p38 MAPK inhibitor (SB203580) result in high XRCC3 mRNA and protein levels (Figure. 4A, 4B). As can be seen in Figure. 4C and 4D, SB203580 reversed the EGCG-induced decrease in XRCC3 mRNA and protein levels in NSCLC cells.
In addition, NSCLC cells were transiently transfected with plasmids containing MKK6E (a constitutively active form of MKK6, which is the upstream protein kinase of p38 MAPK) to determine whether the p38 MAPK signaling pathway was involved in the regulation of XRCC3 protein levels. In NSCLC cells, transfection of MKK6E could promote p38 MAPK activation while lowering XRCC3 mRNA and protein levels (Figure. 4E, 4F). In addition, MKK6E expression dramatically reduced XRCC3 mRNA and protein levels in EGCG-treated NSCLC cells (Fig. 4E, 4F). Our findings suggest that MKK6-p38 MAPK is the upstream signal for reducing XRCC3 protein levels in NSCLC cells.

Figure 4.p38 MAPK activation downregulates XRCC3 mRNA and protein expression in EGCG-treated lung cancer cells. (A, B) NSCLC cells were treated for p38 MAPK inhibitor SB203580 (2.5, 5, and 10 µM) for 24 h. (C, D) NSCLC cells were pretreated for 1 h with the p38 MAPK inhibitor SB203580 (5 µM) before being co-treated for 24 h with EGCG (20 µM). Following treatment, the levels of XRCC3 protein and mRNA in the cell extracts were determined by Western blot analysis and real-time PCR, respectively. (E, F) MKK6E expression vectors were transfected into NSCLC cells. Once the cells had been expressed for 24 h, they were treated with EGCG (40 and 60 M) for 24 h. Total RNA was then extracted, and XRCC3 mRNA expression was determined by real-time PCR (E). Using Student’s t-test, the EGCG-treated cells transfected with the pcDNA3 or MKK6E vectors were compared, **p < 0.01. For the comparison of cells transfected with pcDNA3 or MKK6E vectors, #p < 0.01, using one-way ANOVA. (F) Western blot analysis was used to check the cell extracts after the aforementioned treatment.
p38 mapk activation and downregulation of xrcc3 expression enhanced egcg-induced cytotoxic effects
To investigate the role of p38 MAPK activation in the cytotoxic effect of EGCG, cells were transfected with pcDNA3 or MKK6E expression vectors and then treated with EGCG for 24 h. MKK6E transfection enhanced the cytotoxic and growth-inhibitory effects of EGCG as a means of examining the role of p38 MAPK activation in the cytotoxic effect of EGCG (Fig. 5A, 5B). By contrast, blocking the p38 MAPK activity by SB2023580 could reverse the cytotoxicity and growth-inhibitory effect of EGCG (Fig. 5C, 5D). Furthermore, by using certain siRNA duplexes, XRCC3 mRNA expression was knocked down to ascertain the role of XRCC3 in the cytotoxic effect caused by EGCG. The MTS assay was used to quantify the cytotoxicity induced by EGCG and si-XRCC3 RNA transfection. In Fig. 5E, si-XRCC3 RNA transfection increased the cytotoxicity induced by EGCG. Taken together, both p38 MAPK activation and XRCC3 expression reduction could enhance EGCG-induced cytotoxicity and growth inhibition in NSCLC cells.

Figure 5. p38 MAPK activation and XRCC3 downregulation enhanced EGCG cytotoxicity in NSCLC cell lines. (A) EGCG was applied to NSCLC cells for 24 h after MKK6E (5 µg) or pcDNA3 (5 µg) expression vector transfection. The MTS assay was used to evaluate cytotoxicity. The results (mean ± SEM) were acquired from three independent experiments. (B) After being transfected with pcDNA3 or MKK6E expression vector, the cells were treated with EGCG (20 µM) for 24, 48, and 72 h, and the living cells were counted using the MTS assay. The results (mean ± SEM) were acquired from three independent experiments. ** p < 0>< 0.01, *p < 0.05 (one-way ANOVA) was used. (D) Cells were treated with SB203580 (10 µM) and/or EGCG (40 µM) for 1–3 days, and the living cells were then counted using the MTS assay. The results (mean ± SEM) were acquired from four independent experiments. ** p < 0.01 using the one-way ANOVA to compare cells treated with EGCG alone or GCG and SB203580 combination. (E) The cells were treated with EGCG (20 µM) for 1–4 days following transfection with either XRCC3 siRNA or scrambled siRNA. Living cells were then identified using the MTS assay. Three separate trials were used to generate the results (mean ± SEM). Using Student’s t-test, EGCG-treated cells transfected with either XRCC3 siRNA or scrambled siRNA were compared; **p < 0.01.
Role of XRCC3 and p38 MAPK in EGCG-enhancing erlotinib-inducing cytotoxicity in lung cancer cells
To assess the effect of XRCC3 downregulation on erlotinib cytotoxicity, in erlotinib-treated NSCLC cells, transfection of the si-XRCC3 RNA duplex could drastically reduce XRCC3 mRNA levels (Fig. 6A). In addition, lung cancer cells were treated with EGCG or transfected with si-XRCC3 RNA to suppress XRCC3 expression. EGCG and si-XRCC3 RNA co-treatment could enhance erlotinib cytotoxicity (Fig. 6B). In addition, a p38 MAPK inhibitor was used as pretreatment for NSCLC cells before erlotinib and EGCG administration. The MTS assay was used to measure cell survival. As shown in Fig. 6C, SB203580 could enhance the cell survival that was previously suppressed by EGCG and/or erlotinib. By contrast, the transfection of MKK6E vectors could further reduce cell viability, which is consistent with the cytotoxic effect of EGCG and erlotinib, which was caused by p38 MAPK activation (Fig. 6D). Therefore, EGCG-enhancing erlotinib-inducing cytotoxicity through downregulating XRCC3 expression and activated p38 MAPK in NSCLC cells.

Figure 6. The viability of the EGCG- and erlotinib-induced cells is recovered by inhibiting p38 MAPK activity. (A) Before being treated with erlotinib in complete medium for 24 h, NSCLC cells were transfected with siRNA duplexes (200 nM) targeted to XRCC3 or scrambled (control) in a complete medium. After treatment, cell extracts were used to determine XRCC3 mRNA and protein using real-time PCR. (B) Using lipofectamine, the cells were transfected to express si-XRCC3 RNA. Following 24 h of expression, the cells were treated with erlotinib or EGCG (20 µM) for an additional 24 h, and the MTS assay was used to assess cytotoxicity. c**p < 0.01, using one-way ANOVA for comparison of the erlotinib-treated cells that were transfected with si-XRCC3 or scrambled siRNA. d**p < 0.01, using one-way ANOVA for comparison of the erlotinib-treated or erlotinib and EGCGco-treated cells. (C) NSCLC cells were pretreated with SB2023580 for 1 h and then co-treated with EGCG (10 µM) and erlotinib (10 µM) for 24 h. Cytotoxicity was assessed using the MTS assay. **p < 0.01 using the one-way ANOVA to compare cells treated with EGCG and erlotinib in SB2023580 or DMSO vector-transfected cells. (D) After being transfected with pcDNA3 or MKK6E expression vector, the cells were treated with EGCG and erlotinib for 24 h, and living cells were counted using the MTS assay. The results (mean ± SEM) were acquired from three independent experiments. ** p < 0.01 using the one-way ANOVA to compare cells treated with EGCG and erlotinib in MKK6E or pcDNA3-transfected cells.
Cancer is the world’s most serious challenge for public health in terms of morbidity and mortality. This pathogenesis is caused by several factors, including modifications in several biological processes and cell signaling pathways. Moreover, natural substances or active components of herbs contribute significantly to the improvement of the effectiveness of anticancer treatments and lessen their toxicity. It has been anticipated that pharmacological combinations made of dietary supplements and natural items will produce the same results as traditional chemotherapy with fewer side effects [24]. In this study, EGCG may promote erlotinib cytotoxicity in NSCLC cells, which is linked to a reduction in XRCC3 expression.
Lung cancer, melanoma, breast cancer, leukemia, and colon cancer are a few of the human cancer cell lines that have shown growth suppression and apoptosis when exposed to EGCG in vitro [25, 26]. Lower levels of phospho-c-jun and phospho-ERK1/2 in lung adenomas and carcinomas, decreased cell proliferation, increased apoptosis, and decreased angiogenesis were all linked to the prevention of lung tumor by EGCG in A/J mice [27]. By increasing p53 expression, EGCG is a key factor in the suppression of anchorage-independent development of human lung cancer cells. Moreover, EGCG activity can significantly boost p53 stability and promote p53 nuclear localization [28]. By inhibiting HO-1 expression and enhancing the antitumor effects of EGCG, metformin also amplified EGCG-induced ROS production, which ultimately led to apoptosis [29]. Serum samples from patients with cancer had lower hsa-mir-485-5p levels. Meanwhile, EGCG contributed to the upregulation of hsa-mir-485-5p expression, and hsa-mir-485-5p imitators significantly enhanced cell apoptosis and reduced cancer cell proliferation [30]. Cleaved caspase-3 and Bax were further expressed when EGCG-induced apoptosis was applied to A549 lung cancer cells; however, Bcl-xL was less expressed [31]. Moreover, EGCG effectively reduced lung cancer stem cell activity by lowering lung cancer stem cell markers, preventing the development of tumorspheres, reducing proliferation, and promoting apoptosis [32]. The ability of EGCG to increase interleukin-6 production was explained by its cytotoxic effects on both parental lung cancer cells and their cisplatin-resistant counterparts. However, EGCG did not affect the downstream effector signal transducers of interleukin-6 and activators of transcriptional 3 phosphorylation [33].
The combination of EGCG and curcumin can inhibit tumor growth without causing any serious side effects in the lung cancer xenograft nude mice model [34]. Furthermore, combination of erlotinib and EGCG with classical chemotherapeutics methotrexate or actinomycin-D lead the human placenta choriocarcinoma cells to apoptosis [35]. Our study demonstrated that the combination of EGCG and erlotinib has synergistic cytotoxicity in NSCLC cells through p38 MAPK-mediated XRCC3 downregulation. Taken together, EGCG presents a synergistic effect when co-treated with chemotherapy medicines in different types of tumor cell lines.
Cell apoptosis, differentiation, and death are a few of the crucial physiological processes in which MAP kinases are involved. The p38 MAP kinases, ERK, and c-JNK are the three main MAP kinases found in mammalian cells [25]. Studies have investigated the established role of MAP kinase pathways in EGCG’s control of AP-1 activity. In Ha-ras-transformed human bronchial cells, EGCG treatment has been demonstrated to decrease the phosphorylation c-Jun, ERK1/2, ELK1, and MEK1/2 [36]. Contrary to these reports, EGCG has been shown to significantly boost AP-1 factor-associated responses via a MAP kinase signaling mechanism in normal human keratinocytes, indicating that the signaling mechanism of EGCG activity may differ noticeably in various cell types [37]. In mammalian cells, the JNK and p38 MAPK pathways are parallel MAP kinase cascades [38]. EGCG-induced modulation of MAP kinases may offer fresh approaches for the prevention or treatment of human cancer because the MAP kinase pathway is frequently dysregulated in several malignancies. In rat hepatoma, leukemia, and lung cancer cells, tea catechin EGCG reduces DNA synthesis [25, 27]. Lower levels of phospho-c-jun and phospho-ERK1/2 in lung adenomas and carcinomas, decreased cell proliferation, increased apoptosis, and decreased angiogenesis were all linked to the preventive effect of EGCG on A/J mouse lung tumor [27]. Moreover, the anti-aging gene Sirtuin 1 is important to lung function and the treatment of NSCLC. EGCG has been shown to regulate and inhibit Sirtuin 1 and Sirtuin 1 regulates p38 MAPK. EGCG and erlotinib combined therapy to inhibit XRCC3 expression in NSCLC as a new combined therapy may require the measurement of Sirtuin 1 in these experiments for the long-term survival of patients with NSCLC. Sirtuin 1 activators and inhibitors may determine the EGCG and erlotinib combined therapy [39-41].
To the best of our knowledge, this study is the first to show that EGCG reduces the levels of cellular XRCC3 protein and mRNA in NSCLC cells through the activation of MKK3/6-p38 MAPK. In addition, one of the main elements contributing to the enhancement of erlotinib’s cytotoxic effects is the EGCG-induced down-modulation of XRCC3 protein. As a result, EGCG and erlotinib combined therapy to inhibit XRCC3 expression in NSCLC may be a new combined therapy that can synergistically enhance cytotoxic effects. In vivo animal studies and human clinical trials are needed to confirm that this proposal is effective in enhancing the overall survival of patients with NSCLC without additive side effects.
Not applicable.
The authors declare that they have no conflict of interest.
We thank Dr. Chia-Che Chang and Dr. Jia-Ling Yang for providing us with expression plasmids for transfection.
This study protocol was reviewed and the need for approval was waived by Biosafety Committee and National Chiayi University. The immortalized cell lines used in this study were purchased from the American Type Culture Collection (Manassas, VA, USA). Ethical approval for use of these cells is not required in accordance with local/national guidelines. This study does not contain any studies with human participants or animals performed by any of the authors.
The authors declare that they have no conflict of interest.
This study was funded by grants from the Ministry of Science and Technology, Taiwan, Grant no. MOST 110-2314-B-002 -265 -MY3; MOST 110-2320-B-415-004, and the National Taiwan University Hospital, Hsin-Chu Branch, Taiwan.
Yun-Wei Lin, Jen-Chung Ko and Jyh-Cheng Chen contributed to conception, design, data analysis and interpretation, and drafting the manuscript. Ching-Hsiu Huang, Pei-Jung Chen, Qiao-Zhen Chang, Bo-Cheng Mu, Jou-Min Hsieh, Pei-Yu Tseng, Chen-Shan Chiang, Jun-Jie Chen and Yun-Wei Lin contributed to data acquisition, data interpretation, and critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work ensuring integrity and accuracy.
All data generated or analysed during this study are included in this article. Further enquiries can be directed to the corresponding author.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
