
Research Article | DOI: https://doi.org/10.31579/2693-7247/118
*Corresponding Author: Sahar Jaffal, PhD in Biological Sciences (Physiology and Neuroscience), Jordan.
Citation: Sahar Jaffal, (2023). Effects of Pgf2α On P-Erk1/2 Mapk Activation, Proliferation and Formation of Trans-Endothelial Tunnels in Swiss 3t3 Fibroblast Cells, J. Pharmaceutics and Pharmacology Research, 6(2); DOI:10.31579/2693-7247/118
Copyright: © 2023, Sahar Jaffal. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 08 February 2023 | Accepted: 20 February 2023 | Published: 28 February 2023
Keywords: fibroblasts; proliferation; PGF2α; FP receptor; trans-endothelial tunnels; p-ERK MAPK
Fibroblast cells are key cells that play a pivotal role in maintaining hemostasis in the body. Keeping the balance between proliferation, differentiation and apoptosis of fibroblast cells is fundamental for the role of these cells. Many mediators can increase fibroblasts' proliferation including prostaglandin F2α [PGF2α]-induced phosphorylated extracellular signal regulated kinase [p-ERK1/2] mitogen activated protein kinase [MAPK]. Thus, the effects and mechanisms of PGF2α in p-ERK1/2 MAPK activation, proliferation and the cytoskeleton of fibroblast cells were explored in the current study. The results showed that sustained p-ERK1/2 MAPK activation was dependent on MAP kinase kinase [MEK-1], proto-oncogene non-receptor tyrosine kinase [c-Src], protein kinase C [PKC] and insulin like growth factor-1 receptor [IGF1R]. The transient p-ERK1/2 MAPK activation involved c-Src, phosphatidylinositol 3-kinase [PI3K]/AKT and PKC pathways. Further, Gαq but not Gαi was involved in the activity of PGF2α in inducing p-ERK1/2 MAPK activation at all time points. In contrast, p-ERK1/2 MAPK activation evoked by PGF2α did not involve Rho, Rho-associated protein kinase [ROCK], adenylyl cyclase [AC], the mammalian target of rapamycin [mTOR] or epidermal growth factor receptor [EGFR].
Notably, PGF2α increased fibroblasts' proliferation via PI3K, MEK1, c-Src, PKC and IGF1R but not EGFR, Gαi, AC or mTOR pathways suggesting a correlation between proliferation and pathways that are involved in the transient and sustained p-ERK1/2 MAPK activation or in the sustained phase alone. Additionally, PGF2α produced a noticeable change in the cytoskeleton of fibroblast cells and increased the number of trans-endothelial tunnels by 54.12% compared to control group. These findings provide further insights into the mechanisms of PGF2α in p-ERK1/2 MAPK activation, proliferation and the cytoskeleton of swiss 3t3 fibroblast cells and can open an avenue to conduct more researches on PGF2α signalling to tailor drugs that can prevent specific routes in PGF2α-FP signalling pathways and decrease fibroblasts' proliferation.
Fibroblast cells are principal cells responsible for the production, maintenance of extracellular matrix and tissue microenvironment [1]. Several lines of evidence showed that dysregulation in the balance between differentiation and proliferation of myofibroblast leads to fibrosis and abnormalities in wound healing [1]. Notably, many mediators can increase fibroblasts' proliferation including prostaglandin F2α [PGF2α]-induced phosphorylated extracellular signal regulated kinase [p-ERK1/2] mitogen activated protein kinase [MAPK]. Importantly, earlier reports revealed that swiss 3t3 fibroblast cells express FP receptor whereby prostaglandin F2α [PGF2α] causes phosphoinositide [PI] turnover induced by phospholipase C [PLC] and calcium [Ca+2] pathways [2-4]. Generally, FP receptor belongs to the family of G protein coupled receptors [GPCRs] that are coupled to heterotrimeric G proteins consisting of α and βγ subunits [5,6]. FP receptor couples to Gαq that activates PLC pathway alongside Gα12/13 that stimulates Rho/Rho-associated protein kinase [ROCK] pathway leading to effects on different effectors and responses in swiss 3t3 fibroblast cells [2]. It is also known that the protein kinase C [PKC] plays key role in the signalling of FP receptor [7]. It merits consideration that GPCRs can signal by trans-activation through different receptor tyrosine kinases such as epidermal growth factor receptor [EGFR], platelet derived growth factor receptor and insulin like growth factor-1 receptor [IGF1R] [8,9]. Notably, activation of receptor tyrosine kinases plays a vital role in the proliferation and differentiation of fibroblast cells, thus contribute to important physiological processes such as tumor progression, wound healing and tissue homeostasis. Also, trans-activation can occur via non receptor tyrosine kinases such as the proto-oncogene non-receptor tyrosine kinase [c-Src] and Pyk2 [10,11].
In line of fibroblasts' proliferation, Oga and co-workers shed the light on the role of PGF2α and FP receptor in inducing fibrosis by increasing collagen expression and fibroblasts' proliferation [12]. Moreover, Dekanty and co-workers referred to the role of PGF2α in increasing the proliferation of swiss 3t3 cells by different mechanisms including the increase in ERK phosphorylation [13].
Multiple reports described that PGF2α increases fibroblasts' proliferation via ERK1/2 MAPK but not p38 MAPK [13]. Noteworthy, the strength and localization of MAPK is an important factor in determining the outcome of MAPK activation. In fact, there are many contributors to the duration and strength of ERK MAPK such as receptor density at the cell surface, the interplay between different phosphatases and kinases, the involvement of scaffolding proteins and the extracellular matrix [14]. For example, it was reported that sustained ERK is activated by effectors from the intracellular membrane compartments [e.g., Golgi] but not the plasma membrane [14]. The proximity of these intracellular compartments to the nucleus is responsible for the translocation of sustained ERK MAPK to the nucleus [14]. On the other hand, many studies highlighted the physiological effect of the crosstalk between phosphatidylinositol 3-kinase [PI3K]/AKT survival pathway and the mitogenic pathway [p-ERK1/2 MAPK] in breast cancer cells [15,16]. It is well-recognized that the interaction between PI3K/AKT and MAPK can determine cell fate [15]. Notably, the mammalian target of rapamycin [mTOR] is one of the PI3K-related kinases that act downstream AKT and take part in several biological responses [17]. Moreover, it is well-documented that that there is a correlation between proliferation and changes in cytoskeleton. In more details, Ohno and co-workers showed a link between rapidly proliferating cultures of fibroblasts and organized actin fibers in skin cultures from human [18]. The aim of this work was to determine the pathways that are involved in the transient and sustained p-ERK1/2 MAPK activation that is evoked by PGF2α and to determine the link with proliferation and cytoskeleton. To the best of the author's knowledge, none of the previous studies reported the crosstalk between the PI3K/AKT and p-ERK1/2 MAPK pathways in fibroblast cells following PGF2α treatment despite the plethora of researches published about PGF2α signalling. Therefore, the interplay between the two pathways in fibroblast cells was explored in this research.
2.1 Materials
AL8810 and PGF2α were from Cayman [Ann-Arbor, MI]. Enhanced Chemiluminescence [ECL], [3H] thymidine and ERK1/2 AlphaScreen SureFire kit were purchased from Perkin Elmer [Waltham, MA]. LY294002, PP2, rapamycin and PD98059 were from Calbiochem [St. Louis, US]. SQ22536 was from Tocris Bioscience [Bristol, UK]. C3 exoenzyme was brought from Cytoskeleton [Denver, CO]. Mouse monoclonal phosphorylated ERK1 [Thr202/Tyr204], rabbit polyclonal total ERK [Thr185/Tyr178], phospho-AKT [Ser473, D9E] and AKT [C67E7] rabbit monoclonal antibodies were from Cell Signaling [Danvers, MA]. The antibody of c-Src [GD11] was obtained from Upstate Biotechnology [Bedford, MA]. Dulbecco’s modified Eagle’s medium [DMEM] and phosphate buffered saline [PBS] were provided by HyClone [Logan, UT]. Y27632 was from brought from Abcam [Cambridge, UK]. Lipofectamine 2000, heat-inactivated fetal bovine serum [FBS], pcDNA3.1 were from Invitrogen [Carlsbad, CA]. Dimethysulfoxide [DMSO], trichloroacetic acid [TCA] and sodium hydroxide [NaOH] were from BioShop Canada Inc. [Burlington, Canada]. Anti-mouse secondary antibody, anti-rabbit secondary antibody and pertussis toxin [PTX] were from Sigma-Aldrich [St Louis, US]. AG1478, AG1024, G06983 and bovine serum albumin [BSA] were from EMD Chemicals Inc [Gibbstown, NJ]. c-Src dominant negative [K298R] was described elsewhere [19]. Alexa-Fluor 488 phalloidin antibody was purchased from Molecular Probes [Eugene, OR]. Ethanol, paraformaldehyde, autoradiography films, non-fat dry milk and other materials needed for western blot were ordered from Santa-Cruz Biotechnology [Dallas, USA]. Mounting medium was brought from Thermo Fisher Scientific [Waltham, USA].
2.2 Drug preparation
PGF2α, PD98059, SQ22536, AG1024, AG1478, LY294002, rapamycin, PP2 and AL8810 were dissolved in DMSO. PTX and Y27632 were solubilized in distilled water while C3 exoenzyme was dissolved in a solution of glycerol and distilled water [50% v/v]. G06983 was solubilized in ethanol.
2.3 Cell culture, treatment and transfection
Swiss 3t3 fibroblast cells were maintained as described in Braga et al. [20]. Swiss 3t3 fibroblasts were grown in uncoated 12 well plate at a density of 70000 cells/well in DMEM media containing 10
3.1 Activation of p-ERK1/2 MAPK by PGF2α in swiss 3t3 fibroblast cells
In the present study, the expression of FP receptor was determined by identifying the ability of PGF2α to induce p-ERK1/2 MAPK activation in fibroblast cells. For this purpose, antibodies that recognize p- ERK1 [Thr202/Tyr204] and ERK2 [Thr185/Tyr178] were used. The results showed that the maximum activation of p-ERK1/2 MAPK induced by 1 µM PGF2α was at 2 min [Figure 1A]. This activation decreased but was sustained upon stimulating the cells with PGF2α for 5, 15, 30 and 60 min. Based on the results of this study, the signalling of PGF2α at 2 min was named as a transient p-ERK1/2 MAPK activation while the time points beyond the 2 min were referred to as sustained p-ERK1/2 MAPK activation. Also, PGF2α triggered p-ERK1/2 MAPK in a concentration dependent manner [Figure 1B]. In all cases, the activation of p-ERK1/2 MAPK by PGF2α was inhibited by using the FP selective antagonist, AL8810 [data not shown].
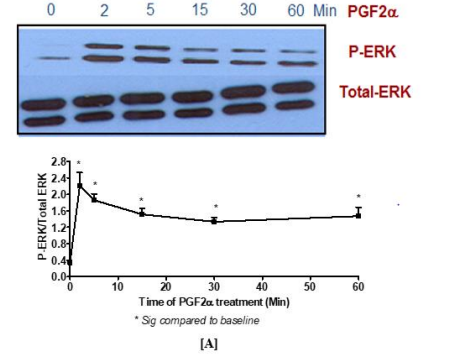
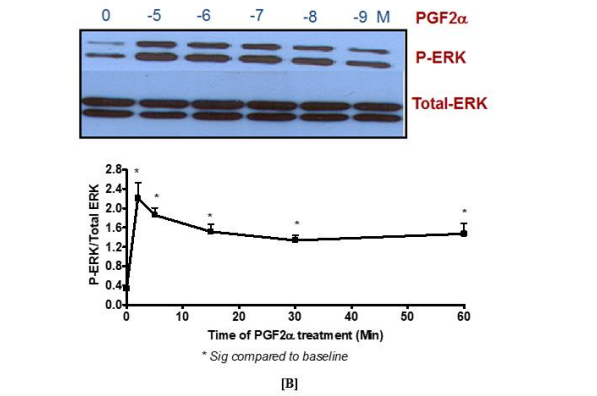
Figure 1. Activation of p-ERK1/2 MAPK by PGF2α in swiss 3t3 fibroblast cells in a time and [A] concentration [B] dependent manner
Data are expressed as Mean±SEM of phosphorylated ERK/total ERK ratio. Western blot is representative of 3 independent experiments.
3.2 PGF2α induces MEK-1 and IGF1R dependent sustained activation of p-ERK1/2 MAPK in swiss 3t3 fibroblast cells
The findings of this study indicated that PGF2α-induced transient p-ERK1/2 MAPK activation [at 2 min treatment] was not inhibited by pre-treatment with the selective inhibitor of MEK1 [PD98059, 50 µM]. In contrast, stimulating fibroblast cells with this inhibitor prior to 1 µM PGF2α treatment for 5-60 min completely abolished the signal of p-ERK1/2 MAPK compared to DMSO-treated cells [Figure 2]. To add, the sustained but not transient p-ERK1/2 MAPK activation evoked by PGF2α was inhibited by using a selective inhibitor of the tyrosine kinase IGF1R [AG1024, 1 µM] indicating the involvement of the transactivation mechanism through this tyrosine kinase in PGF2α-evoked p-ERK1/2 MAPK augmentation in the sustained phase [Figure 3].
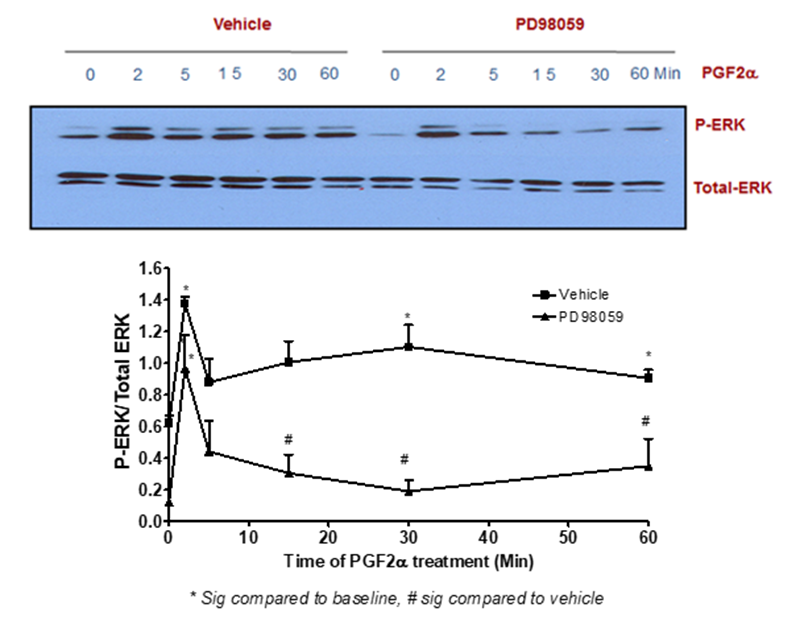
Figure 2: MEK1 inhibition abolished the sustained but not transient PGF2α –mediated p-ERK1/2 MAPK activation in swiss 3t3 fibroblast cells
Data are expressed as Mean±SEM of phosphorylated ERK/total ERK ratio. Western blot is representative of 3 independent experiments
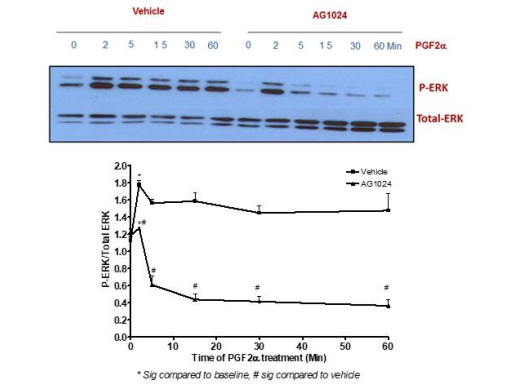
Figure 3: PGF2α-induced increase in sustained p-ERK1/2 MAPK occurs through trans-activation of IGF1R in swiss 3t3 fibroblast cells.
Data are expressed as Mean±SEM of phosphorylated ERK/total ERK ratio. Western blot is representative of 3 independent experiments.
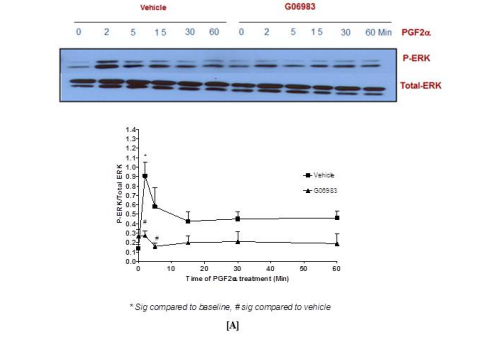
3.3 Determining the type of Gα protein responsible for p-ERK1/2 MAPK activation in PGF2α-FP signaling
To determine the involvement of Gαq and Gαi proteins during the transient and sustained p-ERK1/2 MAPK activation evoked by PGF2α, specific inhibitors for the broad-spectrum PKC [G06983, 1 µM] and Gαi [PTX, 100 ng/ml] were used. The results showed that the transient and sustained p-ERK1/2 MAPK activation were inhibited by pre-treating fibroblast cells with the PKC inhibitor prior to PGF2α stimulation indicating the coupling of FP receptor to Gαq at all time points [Figure 4A]. On the contrary, there was no evidence for the involvement of Gαi in PGF2α-induced elevation of p-ERK1/2 MAPK in either of the phases in swiss 3t3 fibroblast cells [Figure 4B].
[B]
Figure 4: Involvement of Gαq [A] but not Gαi [B] upon PGF2α stimulation in swiss 3t3 fibroblast cells.
Data are expressed as Mean±SEM of phosphorylated ERK/total ERK ratio. Results of Alpha Screen or western blots are representative of 3 independent experiments.
3.4 PGF2α induces an increase in p-ERK1/2 MAPK in the transient and sustained phases independently of many pathways in swiss 3t3 fibroblasts
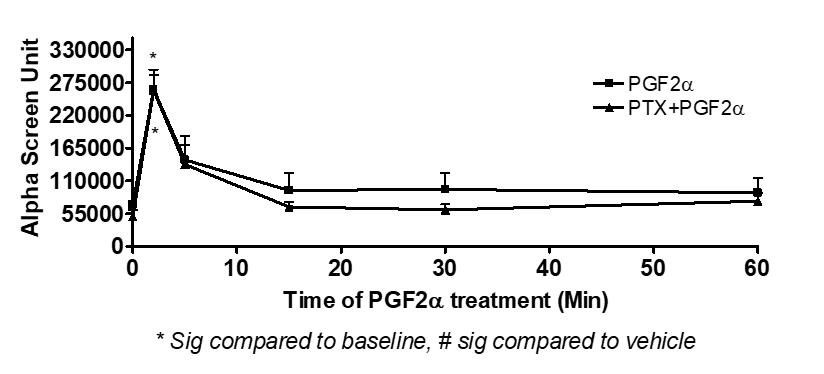
The transient and sustained elevation of p-ERK1/2 MAPK mediated by PGF2α did not involve AC [Figure 5], Rho pathway [Figure 6], ROCK [Figure 7] or the trans-activation through EGFR [Figure 8] in fibroblast cells.
Figure 5: No involvement of AC in PGF2α-induced transient or sustained p-ERK MAPK in swiss 3t3 cells..
Data are expressed as Mean±SEM of phosphorylated ERK/total ERK ratio. Results of AlphaScreen are representative of 3 independent experiments.
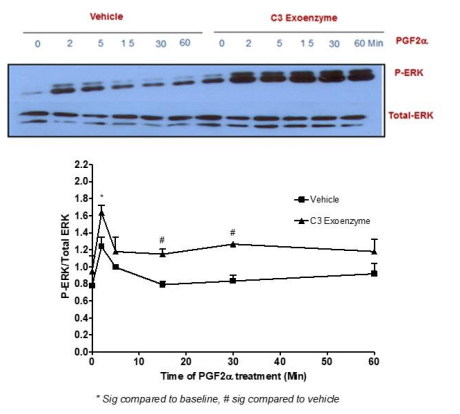
Figure 6: No involvement of the Rho pathway in p-ERK MAPK activation mediated by PGF2α in swiss 3t3 fibroblasts.
Data are expressed as Mean±SEM of phosphorylated ERK/total ERK ratio. Western blot is representative of 3 independent experiments.
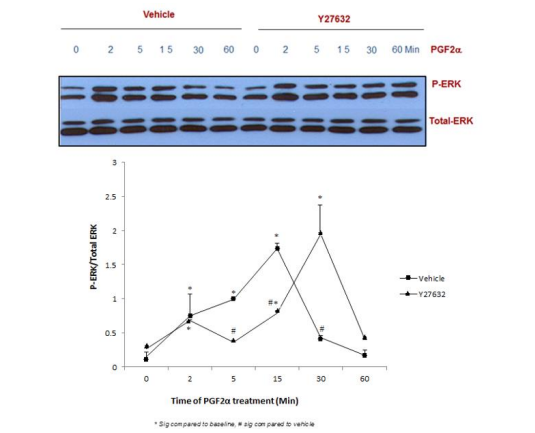
Figure 7: Blocking ROCK pathway did not affect p-ERK1/2 MAPK mediated by PGF2α in swiss 3t3 fibroblasts.
Data are expressed as Mean±SEM of phosphorylated ERK/total ERK ratio. Western blot is representative of 3 independent experiments.
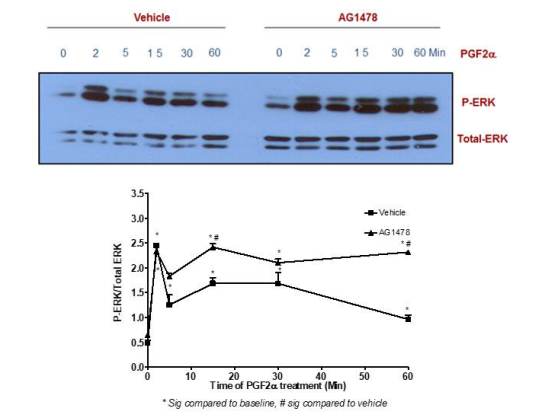
Figure 8: Blocking EGFR did not affect p-ERK1/2 MAPK mediated by PGF2α in swiss 3t3 fibroblasts.
Data are expressed as Mean±SEM of phosphorylated ERK/total ERK ratio. Western blot is representative of 3 independent experiments.
3.5 Elucidating the interaction between the survival and mitogenic pathways in swiss 3t3 fibroblasts
The effectors in the survival pathway and their involvement in PGF2α induced p-ERK1/2 MAPK activation in fibroblast cells were examined using antibodies that detect phosphorylated ERK1 and ERK2 and the phosphorylation of AKT on Ser473 residue. Also, a reversible inhibitor for PI3K [LY294002] was added to the cells prior to PGF2α treatment. The findings demonstrate the involvement of PI3K in activating p-ERK1/2 MAPK at 2 min of PGF2α treatment [Figure 9A]. The effectiveness of the PI3K inhibitor was tested by blotting the membranes with total AKT antibody [Figure 9B]. Additionally, the involvement of a PI3K related kinase [mTOR] in PGF2α induced p-ERK1/2 MAPK activation was explored using rapamycin inhibitor. No change in p-ERK1/2 MAPK elevation was revealed in the group that was pre-treated with rapamycin prior to PGF2α compared to vehicle treated group in the two phases of activation [Figure 9C].
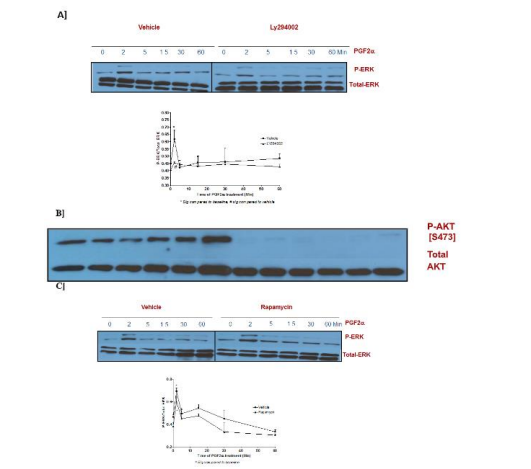
Figure 9: Interaction between the survival PI3K/AKT and the mitogenic ERK1/2 MAPK pathways in PGF2α treated fibroblast cells.
Data are expressed as Mean±SEM of phosphorylated ERK/total ERK ratio. Western blot is representative of 3 independent experiments.
3.6 PGF2α enhances p-ERK1/2 MAPK activation through c-Src activation
To test the involvement of c-Src in the signaling of PGF2α in swiss 3t3 fibroblast cells, the cells were treated with c-Src family kinase inhibitor [PP2, 10 µM] for 30 min followed by 1 µM PGF2α treatment for different times. Suppression of c-Src by PP2 diminished p-ERK1/2 MAPK activation mediated by PGF2α in the transient and sustained phases of p-ERK1/2 MAPK activation in swiss 3t3 cells [Figure 10A]. To confirm this result and because of the non-selectivity of PP2, the cells were transfected with a kinase inactive mutant of c-Src [K298R] or its vector [pcDNA3.1] in swiss 3t3 fibroblasts. The results of transfection were in agreement with the findings of using PP2 inhibitor. There was a noticeable reduction in p-ERK1/2 MAPK activation in the group that was pre-transfected with dead mutant of c-Src [Figure 10B] compared to pcDNA3.1-transfected cells indicating the involvement of c-Src in p-ERK1/2 MAPK stimulation evoked by PGF2α in swiss 3t3 in both phases.
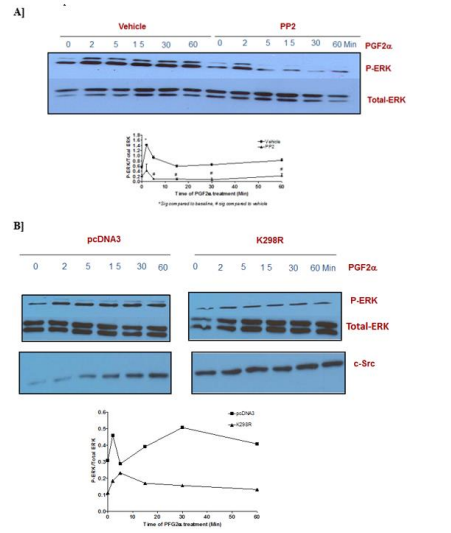
Figure 10: Involvement of c-Src in the transient and sustained ERK1/2 MAPK activation in fibroblast cells.
Data are expressed as Mean±SEM of phosphorylated ERK/total ERK ratio. Western blot is representative of 3 independent experiments.
3.7 Summary of the different pathways that are involved in the transient and sustained ERK MAPK activation in swiss 3t3 cells
Figure 11A and 11B represent schematic representations for the pathways that are involved in PGF2α-evoked transient and sustained p-ERK1/2 MAPK activation in fibroblast cells, respectively.
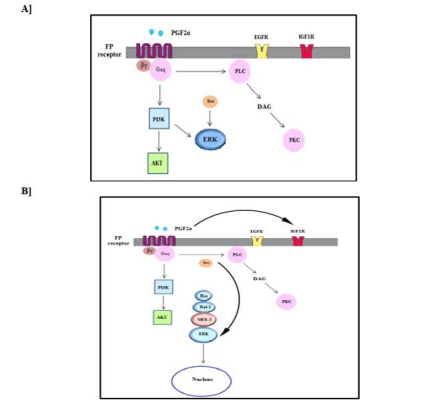
Figure 11: Diagram of the pathways involved in the transient [A] and sustained [B] ERK MAPK activation evoked by PGF2α in fibroblast cells.
3.8 PGF2α enhances fibroblasts' proliferation in a concentration dependent manner via PI3K, MEK-1, PKC, c-Src and trans-activation through IGF1R pathways
The findings of this study revealed that PGF2α increased fibroblasts' proliferation percent in a concentration dependent manner compared to control group [Figure 12A]. PGF2α [1 µM] was selected to test the effect of different inhibitors on PGF2α-enhanced proliferation in fibroblast cells, because this concentration was effective in increasing proliferation and is similar to the concentration that was used in western blot experiments. It was found that PGF2α-induced increase in proliferation was inhibited by the pre-administration with PI3K, MEK1, PKC, c-Src and IGF1R inhibitors. Meanwhile, the inhibitors of Gαi, AC and mTOR didn't affect PGF2α-mediated proliferation [Figure 12B].
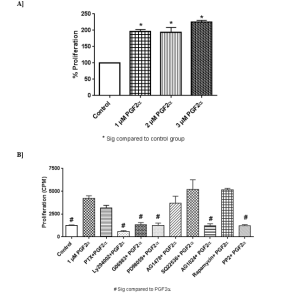
Figure 12: Effect [A] and pathways [B] of PGF2α in swiss 3t3 fibroblasts' proliferation.
3.9 PGF2α increases the percentage of trans-endothelial tunnels in fibroblast cells compared to control group
To explore the effect of PGF2α on F-actin and cytoskeleton, Alexa-Fluor 488 phalloidin labeling and confocal microscopy were used. Table 1 presents the number of cells with or without trans-endothelial tunnels in different treated groups of cells. Compared to control group [Figure 13A], the cells treated with 1 μM PGF2α had more trans-endothelial tunnels [Figure 13B]. Interestingly, fibroblast cells clearly appeared as perforated cells and included tunnels with different diameters. In more details, the number of PGF2α-treated cells that have trans-endothelial tunnels increased by 40% compared to non-treated cells. This finding indicates that PGF2α induced changes in actin and cytoskeleton. Moreover, neither AC inhibitor nor Gαi inhibitor caused a significant change in the percentage of trans-endothelial tunnels compared to PGF2α-treated group [Table 1]. However, there was a noticeable enlargement in the diameter of trans-endothelial tunnels upon treating the cells with AC inhibitor [Figure 13C] but not Gαi inhibitor [Figure 13D].
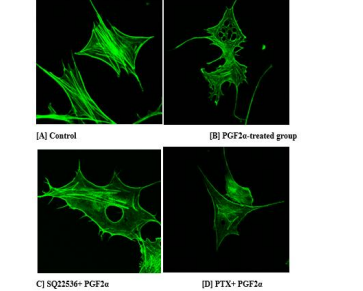
Figure 13: Representative images for the trans-endothelial tunnels-induced by PGF2α in fibroblast cells compared to control group.
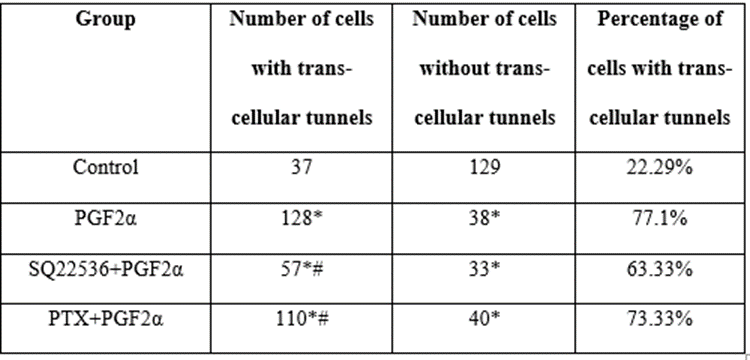
* Sig compared to control group, # Sig compared to PGF2α-treated group
Table 1: Number of cells with or without tunnels in different treated groups.
Fibroblasts' proliferation is one of the characteristics that occur during fibrosis. It is well-established that there is a correlation between proliferation, PGF2α and p-ERK1/2 MAPK activation in fibroblast cells and a role for FP receptor in fibrosis [12, 13, 21]. In more details, it was noted that sustained prolonged ERK1/2 activation is responsible for increasing proliferation in fibroblast cells [22]. Moreover, there is an important role for PGF2α in keeping the balance between fibroblasts' proliferation and differentiation. Thus, the aims of this research were to determine the effects PGF2α in p-ERK1/2 MAPK activation, proliferation and cytoskeleton in swiss 3t3 fibroblast cells that express FP receptor and unravel the mechanisms that underlie this effect.
In the current study, upon PGF2α stimulation, it was found that the highest peak of p-ERK1/2 MAPK activation was at 2 min [transient activation]. This signal decreased and remained stable after the 2 min treatment [sustained activation] indicating that different mechanisms are involved in PGF2α-induced p-ERK1/2 MAPK activation in the transient and sustained phases.
Importantly, previous studies attributed the difference in the strength and the duration of p-ERK1/2 MAPK activation to several factors including the localization of p-ERK1/2 MAPK and the activation of phosphatases [14]. In this regard, it was illustrated that the phosphorylation of ERK MAPK in response to mitogens is associated with p-ERK translocation to the nucleus and the phosphorylation of transcription factors that take part in proliferation [e.g. c-Fos]. However, the activated p-ERK can remain cytoplasmic, in some cases, leading to different effects depending on the type [23], age [24] and function of the cell [25]. Furthermore, the difference in biological outcomes of the transient or sustained ERK1/2 activation is related to the activation of genes such as c-Fos [14]. In more details, c-Fos can be phosphorylated by sustained active ERK and other kinases leading to many effects that are not involved in the transient ERK activation [14].
In support of this contention, Volmat and co-authors revealed that sustained ERK activation is associated with signal localization to the nucleus where it can be dephosphorylated by phosphatases in the nucleus but not the cytoplasm such as MAPK phosphatases1/2 [26]. Accordingly, it is expected that the transient p-ERK1/2 MAPK that was activated by PGF2α is cytosolic whereby there is lack of many phosphatases in the cytoplasm in contrast to the nuclear sustained p-ERK1/2 MAPK activation that is exposed to a reduction in the signal by phosphatases. Another explanation for the stronger signal in the transient phase observed in this work is that the time of two minutes is a short period that is not sufficient for the degradation of p-ERK1/2 MAPK activating factors through the proteasome compared to longer treatments. These differences in the mechanisms of transient and sustained stimulation of p-ERK1/2 MAPK can lead to variations in the expression of many genes, translocation of ERK to the nucleus and can be translated to differences in physiological effects and cell fate [e.g. proliferation or differentiation] [14; 27]. Moreover, the results showing that the activation of transient p-ERK1/2 MAPK by PGF2α was mediated by PI3K can be explained by the fact that AKT activation during the transient phase suppresses glycogen synthase kinase 3β that positively regulates multiple protein phosphatases [MKPs] responsible for ERK dephosphorylation [28]. This finding adds another possible explanation for the maximum peak of ERK that was observed at 2 min PGF2α stimulation in this study.
Despite the fact that MEK1 is the upstream kinase that phosphorylates ERK in two conserved residues, threonine and tyrosine [15], the data of this study indicate that PGF2α-induced p-ERK1/2 MAPK activation in the transient phase was not inhibited by the pre-treatment with the selective inhibitor of MEK1. This result suggests that PGF2α-induced p-ERK1/2 MAPK transient activation was resistant to the canonical pathway of p-ERK1/2 MAPK activation that involves MEK1 and may be to MEK1 upstream effectors. In contrast, MAPK activation in the sustained phase was MEK dependent.
In fact, the presence of PKC in PGF2α signaling at all-time points including the transient phase [which is MEK1 independent] raised a question regarding the explanation of this finding as PKC is well known to phosphorylate MEK1 leading to ERK1/2 activation. Several studies highlighted the fact that there are many routes whereby PKC can trigger p-ERK1/2 MAPK such as the indirect phosphorylation of ERK by PKC through raf or MEK1 [29-31]. Other routes include direct PKC phosphorylation without involvement of ras or MEK1 [29-31]. Accordingly, the activation p-ERK1/2 MAPK by PKC or other mechanisms is not necessarily to be mediated through ras-raf-MEK-ERK pathway [32]. In addition, there are many types of PKCs and not all of them depend on MEK1 [33]. For instance, the constitutive active PKCµ depends entirely on MEK activity but the other types of PKC can be activated by different routes [33]. Thus, it is expected that the type of PKC triggered in the transient phase in the present study is not similar to the other types of PKC kinases recruited at other time intervals.
Generally, the differences in the signalling of GPCRs can be attributed to their ability to switch G proteins that lead to recruitment of different effectors and signaling pathways [5-6]. As expected, the results in this work confirmed the involvement of Gαq in the signalling of FP receptor in swiss 3t3 fibroblast cells during the transient and sustained p-ERK1/2 activation. Importantly, the possibility of the involvement of Gαi in PGF2α-mediated effects in fibroblast cells was tested for many reasons. First, Hébert et al. pointed to the Gαi recruitment by PGF2α in rabbits' kidney [34]. Second, it was reported that PGF2α increased the phosphorylation of cyclic adenosine monophosphate [cAMP] response element binding protein 1 [CREB1] via FP receptor and induced PKC activation in amnion fibroblasts [7]. Also, other studies indicated the production of cAMP by PGF2α in bovine iris sphincter smooth muscle [35]. Importantly, there was no involvement of Gαi in PGF2α induced p-ERK1/2 MAPK in fibroblast cells in either phase [transient or sustained p-ERK1/2 MAPK activation] in this study. Finally, to the best of the author's knowledge, none of the previous reports studied the recruitment of Gαi by FP receptor in swiss 3t3 fibroblasts.
As an alternative mechanism, accumulating lines of evidences revealed that many GPCRs induce their signal via the trans-activation through receptor tyrosine kinases such as EGFR and IGF1R. In this work, there was no inhibition of p-ERK1/2 MAPK signal at any time point in the group that was pre-treated with EGFR inhibitor in swiss 3t3t cells. In contrast, the activation of p-ERK1/2 MAPK by PGF2α involved the trans-activation through IGF1R during the sustained p-ERK1/2 MAPK activation. Notably, IGF1R is a trans-membrane tyrosine kinase that activates PI3K through the insulin response substrate-1 [IRS1] [36]. Notably, p-ERK1/2 MAPK at the sustained phase wasn't affected by the inhibition of PI3K which is known to be activated by IGF1R. Accordingly, there is possibility that PGF2α has a direct inhibitory effect on IRS1 and its ability to couple efficiently to PI3K through phosphorylation of its serine residues [37-39]. Other explanation is that IGF1R can cause a negative feedback loop that induces the phosphorylation of serine residues of IRS1 [37]. It could be a good area for future work to examine if the involvement of PI3K in the activation of PGF2α-induced p-ERK1/2 MAPK is mediated by direct inhibition of AKT or by the inhibition of AKT upstream regulators of AKT such as Grb2 associated binder [GAB1], phosphoinositide dependent kinase 1 [PDK1] and p21 activated kinase 1 [PAK1]. Further, c-Src was tested by using an inhibitor for c-Src and a kinase dead mutant of c-Src [K298R] as c-Src is one of the pathways that several GPCRs can signal through [10-11]. Our results showed that the involvement of c-Src in the transient and sustained phases. Noteworthy, the author examined the involvement of Rho/ROCK pathway in PGF2α mediated elevation in p-ERK1/2 MAPK for two reasons. First, most of the receptors that couple to Gαq are able to couple to Gα12/13 which activates Rho/ROCK pathway [6]. Second, Ding and associates reported that PGF2α increased collagen synthesis in cardiac fibroblast cells via PKC and Rho pathways. In the present study, there was no role for Rho or ROCK in the activation of p-ERK1/2 MAPK [40]. There is possibility for the involvement of other kinases that act downstream AKT such as the novel kinase T-LAK cell-originated protein kinase [TOPK] that was involved in the positive regulation of p-ERK1/2 MAPK by the survival pathway in different cancer cells in MEK1 independent manner [33, 41].
Notably, the pathways that were exclusively involved in transient but not sustained p-ERK1/2 MAPK activation were not implicated in PGF2α-induced proliferation. The findings of previous researches provide a solid justification for these results whereby sustained ERK activation was associated with the entry to the synthesis phase [S phase] of the cell cycle and accordingly, increase in the proliferation of cells [42]. This occurs through the effect of sustained ERK activation of the expression of several genes. Taken as a whole, the aim of this study was to gain a better understanding for PGF2α-FP pathways in the temporal activation of p-ERK1/2 MAPK, fibroblasts' proliferation and to assess whether there is an effect of PGF2α on the cytoskeleton of swiss 3t3 fibroblast cell.
In addition to the relationship between p-ERK1/2 MAPK and proliferation, a correlation between proliferation and changes in actin was reported previously in fibroblasts [18]. Therefore, the morphology of actin fibers in phalloidin-stained fibroblast cells was examined after PGF2α treatment. Interestingly, PGF2α induced more trans-endothelial tunnels accompanied with bigger sizes. Earlier reports showed that the tunnels' formation was noticed in the cells that were treated with bacterial toxins and edema toxins in a mechanism that includes AC and its effectors [43]. Therefore, two selective inhibitors for AC and cAMP pathways were used to address the role of this pathway in PGF2α-induced tunnel.
Finally, the main objective of this study was to determine the pathways that are involved in PGF2α induced proliferation in fibroblasts. It was found that PGF2α increased fibroblasts' proliferation in a concentration dependent manner; in agreement with previous studies [13].
Interestingly, the pathways that were not implicated in the transient and/or sustained PGF2-evoked p-ERK1/2 MAPK activation [e.g. AC, ROCK, EGFR, Gαi] were not involved in PGF2α-enhanced proliferation in fibroblast cells. Taken as a whole, there are shared pathways between proliferation and elevation in p-ERK1/2 MAPK [transient or sustained phases] mediated by PGF2α.
This study highlighted the pathways that underpin PGF2α-induced p-ERK1/2 MAPK activation, proliferation and cytoskeletal changes in fibroblast swiss 3t3 cells. Interestingly, it was found that the routes that are involved in PGF2α-induced transient and sustained p-ERK1/2 MAPK activation or in the sustained phase alone significantly take part in fibroblasts' proliferation. Further, PGF2α increased the number of trans-endothelial tunnels in fibroblasts indicating that it has significant effect on the cytoskeleton of these cells. These findings can open a path to conduct more researches on PGF2α signalling to tailor drugs that can decrease fibroblasts' proliferation.
The author gratefully acknowledges Maram Jaffal for drawing the diagrams in this paper.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
None
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
