
Research Article | DOI: https://doi.org/10.31579/2766-2314/027
Head of the Protein Research Department, Genetic Engineering and Biotechnology Research Institute, City of Scientific Research and Technological Applications, Universities and Research Center district, New Borg El-Arab, Egypt.
*Corresponding Author: Amro Abd Al Fattah Amara, City of Scientific Research and Technological Applications, Universities and Research Center District, Egypt.
Citation: Amro A. Amara (2021) Different simple protocols for separating different types of coexisted plasmids in the same cell. J, Biotechnology and Bioprocessing 2(3); DOI: 10.31579/2766-2314/027
Copyright: © 2021, Amro Abd Fattah Amara, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited
Received: 24 January 2021 | Accepted: 27 January 2021 | Published: 19 February 2021
Keywords: plasmid, coexisted, isolation, competent cells, transformation efficacy
The existence of mixed plasmids in the same cell is tricky and there is a need for separating them from each other. However, isolating two existed plasmids might be difficult, particularly if they are same in their sizes, with same antibiotic marker, or different only in one or more mutants without different restriction cut. Two different plasmids in the same cells is a natural phenomenon as well as a normal practice in molecular biology experiments. For example during random mutagenesis experiment for a single gene (existed naturally in an operon) using a mutator strain like E. coli XL1 Red, the single mutated gene is then complemented with the other essential genes for producing certain products. Another example, during site directed mutagenesis experiment using double antibiotics selection method, in many cases, the original plasmid is existed side by side with the one carry the new mutant. There are many examples where plasmids coexisted with each other either naturally or under experimental conditions. The problem is how one could separate those plasmids particularly when they are similar in their molecular weight and have the same marker. This study introduces two main strategies; the first is based on increasing or decreasing the competent cells transformation efficacy. Where, in general harvesting competent cells either E. coli or other bacterial strains in the first 2-3 hours (or less) of their cultivation and using the enhanced protocol for competent cells preparation will improve the transformation processes. Letting cells to be more ages will reduce the transformation processes. Using four 2-3 hour grown competent Azotobacter sp enable plasmid transformation. The second strategy for separating the coexisted plasmid is based on using diluted plasmids. The antibiotic screening method is based on blind selection where growing on plat containing the first antibiotic and non growing in the second antibiotic means that the tow existed plasmids are separated. In case of existing of plasmids with the same size and the same antibiotic marker for example during the site-directed-mutagenesis protocol (mutants did not have different restriction enzymes cut), the plasmid is diluted and transformed in recombinant E. coli and each clone was cultivated alone and the mutated region is sequenced. The presence of a single base pair in the site of the mutant means presence of a single plasmids and vice versa. As a conclusion same plasmids with point mutation are usually coexisted. In some cases the coexisted plasmids are with similar antibiotic marker, no different restriction enzyme cut sites are existed, no white and blue selection or any other phenotype for selection. In such cases and similar ones diluting plasmid and transforming them in conditions enable single plasmid per cell must be controlled by the sequencer. The protocols included in this study are summarized from the experiences with random and site directed mutagenesis experiments where plasmid with a single mutant is coexisted with the wild mother plasmid or with the other coexisted different mutants.
Molecular biology tools are in need always for simplifying the used protocols. New idea could reduce time and costs. Site directed mutagenesis is one of the most perfect tools to introduce mutants in genes[1]. The random mutagenesis is another tool for mutagenesis E. coli XL1Red is a mutator strain deficient in three of the primary DNA repair pathways. The mutS (error-prone mismatch repair)[2], mutD (deficient in 3´-to 5´- exonuclease of DNA polymerase III) [3] and mutT (unable to hydrolyse 8-oxodGTP) mutations were present in strain XL1-Red (Stratagene, LaJolla, USA). This strain is able to mutate a gene carried on plasmid and transformed to it. The mutants could be accumulated. From one colony many mutants could be obtained [4]. In nature plasmids are coexisted in a single microbe. This study introduces simple solutions for separating two existed similar or different plasmids in one cell.
E. coli strain
XL1-Blue, recA1, endA1, gyrA96, thi-1, hsdR17 (rK-,mK+), supE44, relA1, λ-, lac [F, proAB, lacIq, (lacZ)M15, Tn10(Tcr)] [5]
Cultures containing the different respective plasmids were grown in 20 ml LB medium with the appropriate antibiotics at 37°C for overnight. 1.5 ml of culture broth in Eppendorf tube was centrifuged at 4,000 rpm for 3 min (Biofuge 13, Heraeus instrument Christ, Osterode), the supernatant was removed. 100 µl ice-cold GET solution was added to the pellet and was completely suspended, followed by addition of 200 µl fresh-prepared SDS-NaOH solution. Clear solution was obtained after gently inverting the Eppendorf tubes up and down, indicating that cell lysis was complete. The protein was precipitated by addition of 150 µl potassium acetate solution. After centrifugation at 13,000 rpm for 15 min (Biofuge A, Heraeus Christ, Osterode), the supernatant was transferred to a new Eppendorf tube, then 900 µl of ice-cold ethanol was added and the pellet was washed with 70% (v/v) ethanol. The pellet was dried either at room temperature or in the vacuum drier. The dried plasmid DNA was dissolved in 10-20 µl RNase-containing TE-buffer or distilled water and stored at -20°C. GET-solution: 25 Mm Tris/HCl, pH 8.0; 10 mM EDTA; 50 mM Glucose. SDS-NaOH solution: 200 mM NaOH; 1% (w/v) SDS. 5 M potassium acetate-solution: 29.5 ml Acetic acid; 100 ml H2Obidest; Adjust the pH to 4.8 with KOH. RNase-solution: 150 mM NaCl, pH 5.0; 1 % (w/v) RNase A (80 °C, 15 min to inactivate DNase); TE-Puffer: 10 mM Tris/HCl ; 1 mM EDTA, pH 8.0 ; TE buffer containing RNase 1µl RNase solution : 100µl TE buffer [6].
Determination of the plasmid DNA concentration
The purity and concentration of the plasmid DNA solution was determined by measuring the absorption at 260 nm and 280 nm. An extinction E260 = 1 corresponds to 50 µg dsDNA ml-1 and 38.5-40 µg ssDNA ml-1. For evaluation of pure DNA preparation, the following equation could be used: E260 nm / E280 nm = 1.8. During routine work with small volumes of plasmid DNA solutions, the concentration of DNA was estimated by following fluorescence of bands within the agarose gel that had been stained with ethidium bromide. The amount of DNA which was just visible after electrophoresis, staining and photography on the gel was set to 2 ng [6].
Preparation of competent cells by CaCl2 method
E. coli cells was grown in 20 ml LB medium at 37°C until OD600 reached 0.5-0.7. The cells were harvested by centrifugation at 4,000 rpm for 10 min. 5-10 ml CaCl2-Tris solution was added to suspend the cells, followed by incubating this suspension on ice for 20 min. The competent cells were harvested by centrifugation at 4,000 rpm for 10 min (Megafuge 1.0R, Heraeus instrument Christ, Osterode) and resuspended in 2 ml CaCl2 solution. The competent cells were then mixed with DMSO to a concentration of 7% (v/v) or with glycerol to a concentration of 10% (v/v) and aliquots of 200 µl were distributed into Eppendorf tubes. The competent cells were then stored at –70°C. CaCl2 - Tris solution: 50 mM CaCl2, 10 mM Tris/HCI, pH 8.0
Preparation of competent cells for long term storage [7, 8]
The Xl1Blue E. coli strain was cultivated in 50 ml LB medium at 37°C until OD600 reached 0.3. After 10 to 15 min incubation on ice, the cells were harvested by centrifugation (15 min; 4,000 rpm). The cell pellet was then suspended in 18 ml RF1 solution. After incubation on ice for 30 min, the cells were again centrifuged for 15 min at 4,000 rpm (Megafuge 1.0R, Heraeus Christ, Osterode) and suspended in 4 ml RF2 solution. Competent cells were distributed to Eppendorf tubes (200 µl per tube) and stored at –70°C. RF1 solution: 100 mM RbCl, 50 mM MnCl2, 30 mM Potassium acetate, 10 mM CaCl2 . 6 H2O, Adjust the pH to 5.8 with acetic acid. RF2 solution: 10 mM RbCl, 10 mM MOPS, 75 mM CaCl2 . 6 H2O, 15 % (v/v) Glycerol, Adjust the pH to 8.5 with NaOH [7, 8].
Transformation of E. coli cells
200 µl competent cells were mixed thoroughly with 1 µl plasmid DNA to be transferred and incubated for 30 min in an ice bath. During this incubation step, the DNA was adsorbed at the surface of the competent cells. For the uptake of the adsorbed DNA, the cells were heated at 42°C for exactly 90 sec and subsequently incubated on ice for 3-5 min. For regeneration of the cells and for expression of the plasmid encoded antibiotic resistance, 800 µl LB medium was added and the cells were subsequently incubated at 37°C for at least 30 min. For isolation of the recombinant clones, 100 µl aliquots were spread on selective solid medium containing the selective antibiotic and the plates were incubated overnight at 37°C.
DNA sequencing and mutant detection
Standard criteria were used to sequence the targeted gene (the gene data not shown) using LI-COR 4000L (LI-COR, Alabama, USA). The changed base pairs were observed manually.
Separation of coexisted plasmids with different antibiotic markers and similar size
Inoculums from Xl1Blue E. coli which contain two plasmids similar in size but with different antibiotic markers added to 25 ml LB medium in 100 ml flask (2 flasks). For each flask one antibiotic is added with the proper amount. Plasmids were collected from the overnight cultivated E. coli strain at 37oC using the above described alkaline lysis protocol for plasmid isolation where one ml of the overnight culture were centrifuged at 4000 rpm and the pellet is used for the plasmid isolation using the above described alkaline lysis protocol for plasmid isolation [6]. The plasmid pellets after isolation and drying at 37oC were re-suspended in 50 µl of sterile bi-distilled water and then 5 µl were diluted each in one ml of sterile distilled water. 10 µl was used to transform the recombinant E. coli XL1Blue (modified from the above described protocol). Further steps were followed as described above. The cells left to grow for six hr and then 50µl of the culture is spread on LB medium with the first antibiotic LB medium plats. The plates are incubated overnight. Colonies from each plat were selected and re-cultivated in the second antibiotic LB medium plates. The growth on the first antibiotic and the non growth on the second antibiotic mean that plasmids are separated from each other (Figure 1) and vice versa.
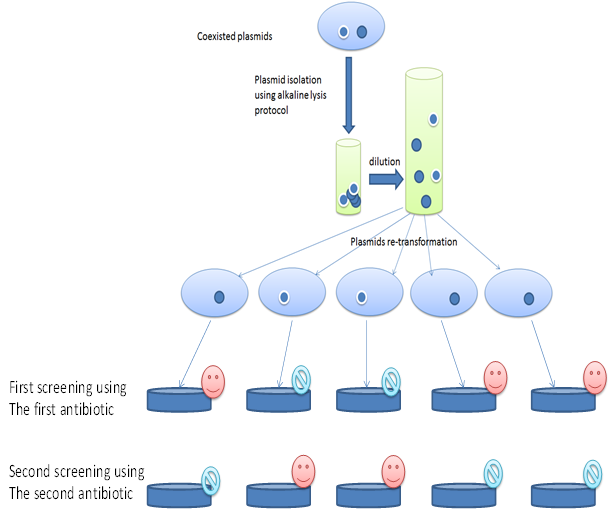
Separation of coexisted similar plasmids with mutated and non mutated genes with similar antibiotic marker
Inoculum from E. coli which contain two plasmids similar in size and in the antibiotic marker but different due to the presence of mutant in the gene is added to 25 ml LB medium in 100 ml flask (one flask). Plasmids were collected from the overnight cultivated E. coli strain at 37oC using the above described alkaline lysis protocol for plasmid isolation [6] where one ml of the overnight culture was centrifuged at 4000 rpm and the pellet is used for the plasmids isolation. The isolated plasmids pellet after drying was re-suspended in 50 µl of sterile bi-distilled water and then 5 µl was further diluted in one ml of sterile distilled water. 10 µl then used to transform the recombinant E. coli XL1Blue. The cells left to grow for six hr and then 50µl of the culture is spread on LB medium with the proper antibiotic plat. Colonies were selected, cultivated and the isolated plasmids from each clone were investigated either by restriction cut if the mutants have different restriction sites to prove the plasmid separation or by using the sequencer to confirm the differences.
Separation of coexisted identical different plasmids with mutants and similar antibiotic marker without restriction cuts
In case of absence of different restriction cut sites, the same process will be followed but instead of using the restriction cut, the sequencing of the mutation region will prove either that plasmid still existed with the wild type or it is already separated. In case of the coexistence of both plasmids the changed base pair will appear in the same line in the sequence image with the original base pair as in Figure 2.

Separation of coexisted plasmids with different antibiotic markers and different sizes
It is recommended to use the recently published fast slot lysis protocol for plasmid isolation as described by Amara (2018) [9]. One can cut the agarose band of each plasmid and use the double Eppendorf tubes centrifugation method for isolating DNA and Plasmids as described by Amara 2018 and 2020 [8, 10].
Using young and old competent cells
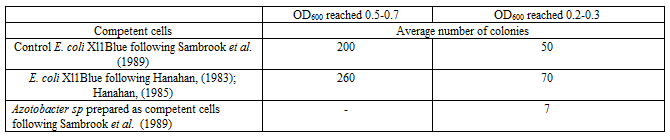
Young (usually 2-3h) OD600 reached 0.2-0.3 or old competent cells (usually 7-10h) OD600 reached 0.5-0.7 either from E. coli or other wild bacterial strains such as Azotobacter sp strain can be prepared for increasing (in case of the young cells) or decreasing of the transformation percentage. Young or old competent cells can be used in separating coexisted plasmid beside the above described tools (Table 1).
This study is a collection of experiences concerning conducting different mutagenesis protocols with different plasmids. In case of random mutagenesis [4] or the site directed mutagenesis [1] using double mutation protocol (where the first mutant is the needed one and the other is caused a second antibiotic resistance) and the selection will be by using a second antibiotic. In some cases different plasmid could be coexisted in the same cells which will be so tricky. One could find two base pairs in the same line in the sequencer image or one could sequence plasmids with (for example) three mutant in the targeted gene and after that one mutant is lost. Those examples represent that different plasmids are coexisted in the same cells. Additionally, in case of sub cloning of a gene in another plasmid similar to the original one or cloning a new gene by eliminating the original gene in the same plasmid or using plasmid to perform PCR amplification for sub-cloning or ligating the amplified gene in new plasmid; in those all cases and more the original plasmid is still existed in the background with few amount but enough to disturb the results. It is recommended to use those described plasmid re-isolation steps to be sure that only one type of plasmid is existed in the used cells.
Separation of coexisted plasmids with different antibiotic markers and similar size
The described method will enable the plasmids separation. The method could be enhanced by adding 25 percentage of the proper antibiotic just after the first hr of the transformation and cultivation steps. That will improve the separation and the selection on the LB medium with the expected antibiotic (data not shown). That also could be reconfirmed by the result of the recultivation on the second antibiotic plat (Figure 1). The growth in the first plate (contain the first antibiotic) and the absence in second one (containing the second antibiotic) using the same colony prove that it contains a single plasmid and vice versa. There is no need for further prove (Figure 1).
Separation of coexisted plasmids with different genes equal in size and similar antibiotic marker
In case that a restriction cut site is existed, cutting with the proper restriction enzyme will prove the presence of a single or different plasmids.
Separation of coexisted identical plasmids with mutants and similar antibiotic marker without restriction cut
In this case the described method will lead to separate the coexisted plasmids but there is a need for sequencing the region where the mutant is designed (in case of the site-directed-mutagenesis). And the whole gene must be sequenced in case of the random mutagenesis. One should remark that to prove the presence of a correct mutant the whole gene must be sequenced from its forwarded and reverse sites.
Separation of coexisted plasmids with different antibiotic markers and different sizes
This might be tricky because the existence of different antibiotic marker will not guarantee separation in case of using a single antibiotic. One antibiotic could cause selection for tranforments containing the different plasmids. However, one could separate the plasmids based on their sizes using the fast slot lysis protocol as described by Amara (2018) [9]. And the plasmids must be separated using the agarose gel electrophoresis. One can use the double Eppendorf tubes centrifugation method to isolate the plasmid from the cut agarose gel bands [10]. The presence of the plasmids could be measured spectrophotometrically as described by Amara (2020) [6, 10].
Using young and old competent cells
Using young competent cells is a wonderful tool for transforming different microbes and can be used for some extend as an alternative to the Electroporation. Young (cells 2-3 hr cultivation OD600 reached 0.2-0.3); was used successfully to transform Azotobacter sp with certain plasmid [data not shown]. Young competent cells of bacteria (other than E. coli) show increase in the number of transforments based on the type of the original cells. In contrast old competent cells show decrease in the number of the transforments and can be used to isolate different coexisted plasmid if diluted. Based on the used E. coli competent cells or other bacteria prepared as competent cells, their age, the competent cells preparation methods and the plasmid purity the percentage of the transformation could be different in each different case (Table 1).
Using different competent cells preparation protocols
Beside the above described tools the type of the method used for competent E. coli preparation will give different results. Using competent cells prepared by CaCl2 method following Sambrook et al. (1989) [6] is recommended due to its low cost and easy handling as described above. Another method proves to be efficient is the competent cells prepared for long term storage as described by Hanahan, (1983) and Hanahan, (1985). It is recommended to use highly transformed protocol [7, 8] in case of very rare plasmids such as in case of the site directed mutagenesis plasmids to guarantee that the correct targeted plasmids are transformed. Then it is recommended to allow the harboring cells (e.g. E. coli) to grow first then start to investigate your clone or separate coexisted plasmid. One should remark that the decrease in the number of colonies in case of XL1Blue at OD600 reached 0.2-0.3 did not mean low transformation efficacy but that due to the presence of lower number of the cells used in the transformation step. However, in case of the Azotobacter sp only at OD600 reached 0.2-0.3 show successful transformation. One should observe the difference effect of the competent cells prepared as young have high transformation % but their low number reduce the total number of the transformants. That might be clearer in case of using Azotobacter sp., where large number but old cells show no transformation as in Table 1.
This study introduces different tools for isolating coexisted plasmids either totally similar such as in case of using plasmids in performing site directed mutagenesis and random mutagenesis without any existence of restriction cut in the new mutated gene. In many cases the sequencer proves that the wild type still existed where the new mutant appear in the same line of the wild type gene. The original base pair and the mutated one appear in the same line. In such cases and the other described ones in this study or those non described there is a real need to separate the coexisted plasmids without losing them. The different experiments introduced in this study will enable the isolation of both of the similar or the different plasmids using two main strategies, the first by reducing the transformation efficacy and the other by diluting the plasmids before their transformation and conducting a transformation experiments guarantee transforming one plasmid/cell.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
