
Research Article | DOI: https://doi.org/10.31579/jcpmh.2021/015
1Orthopedic Hospital of Speising, Pediatric Department, Vienna, Austria
2Family Medicine Operations, Omar Bin Al Khatab Hospital, Doha, Qatar
3Department of Pediatrics, Kazan State Medical University
4Pediatric Orthopedic Institute N.A. H. Turner, Department of Foot and Ankle Surgery, Neuroorthopaedics and Systemic Disorders, Parkovaya Str., 64-68, Pushkin, Saint-Petersburg, Russia
5National Medical Research Center for Traumatology and Ortopedics n.a. G.A. Ilizarov, Kurgan, Russia
6Institute of Human Genetics-Mongi Slim Hospital, Tunis, Tunisia
7Pediatric Orthopedic Surgery, Children Hospital, Tunis
8Department of Medical Laboratory Sciences, Jordan University of Science and Technology, Irbid, 22110, Jordan
9Center of Pathobiochemistry and Genetics, Medical University of Vienna, Austria
*Corresponding Author: Ali Al Kaissi, Orthopedic Hospital of Speising, Pediatric Department, Vienna, Austria.
Citation: Ali Al Kaissi, Marwa Hilmi, Svetlana Volgina, Vladimir Kenis4, Sergey Ryabykh. et all (2021) Desbuquois Syndrome: Misleading Phenotypic Manifestations. J. Clinical Pediatrics and Mother Health, 1(1);Doi: 10.31579/jcpmh.2021/015
Copyright: © 2021 Ali Al Kaissi, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 27 September 2021 | Accepted: 04 October 2021 | Published: 11 October 2021
Keywords: desbuquois type I and 2; phenotype; radiology; CT scan; genotype
Objective: Two groups of children; the first of infants with the preliminary diagnosis of floppy infant syndrome and the second group children diagnosed with congenital scoliosis/kyphoscoliosis.
Material and Methods: Eight patients: six girls and two boys with an age range from 14-months to 14 years were enrolled in this study. Floppy infant syndrome was considered for children born with hypotonia. Scoliosis and kyphoscoliosis were the diagnostic entity for the second group of children. Both groups underwent a detailed and comprehensive clinical and radiographic documentation
Results: Five children manifested the distinctive Desbuquois syndrome type 1 (DBQD-1) and three subjects showed the clinical and the radiological phenotype of Desbuquois syndrome type 2 (DBQD-2). Growth deficiency ranged between (-5SD) to (-7SD). Developmental delay ranged between moderate to global developmental retardation. Early onset cervical kyphosis associated with multiple joint dislocations, overwhelmed by severe growth deficiency of (-7SD) have been encountered in DBQD-1. Late onset scoliosis and angular kyphosis with less articular dislocations were seen more in DBQD-2. A homozygous pathogenic variant in the CANT1 gene was identified in patients with DBQD-1.
Conclusion: Hypotonia associated with prenatal growth deficiency are the first and foremost early features observed in Desbuquois syndrome (type 1). Ligamentous hyperlaxity at birth was misinterpreted by pediatricians and pediatric neurologists as floppy infant syndrome. Placing these infants through endless and prolonged neuromuscular and genetic investigations caused immense torment to their parents. In addition, the time lapsed left a negative impact on the progressive irreversible course of spine tilting. Children with (type 2) Desbuquois manifested the course of spine deformities in their late childhood. The main massage of this paper is to sensitize pediatricians, pediatric neurologists, geneticists and orthopedic surgeons toward the necessity to assess children on the bases of combining clinical features. This requires a better understanding via ameliorating the (clinical- sign -reading power). Two forms of malformation complex have been encountered in this study can be considered as expansion of the skeletal phenotype in DBQD-1. Firstly, Hypoplasia of the big toe seen in all five patients of Desbuquois type 1 may be considered as an expansion of the skeletal phenotype i.e. not reported under the clinical synopsis (OMIM). Secondly, no previous studies described the spine malformation complex as analyzed via comprehensive tomographic findings.
Desbuquois dysplasia (DBQD) is a syndrome of autosomal recessive severe chondrodysplasia and belongs to a group of dysplasias with multiple joint dislocations [1]. It is characterized by facial dysmorphic features, a cleft palate on occasion, pre and postnatal growth deficiency, ligamentous hyper laxity, spine malformations and multiple dislocations [2]. Additional anomalies include talipes equinovarus, metatarsus adductus, rocker bottom feet and so forth [2]. DBQD is radiographically, clinically, and genetically heterogeneous, it is classified into two types according to the presence or absence of distinctive hand anomalies ranging from extra metacarpal ossification center distal to the second metacarpal, delta shape phalanx, or bifid distal thumb phalanx to dislocation of the interphalangeal joints [3]. Developmental retardation has been observed in children manifesting (type -1 Desbuquois) [DBQD-1 (MIM 251450), which is characterized by hyperphalangy of the index finger and (type 2 Desbequois) [DBQD-2 (MIM 615777) in children manifesting the advanced carpal ossification. DBQD-1 seems to be a complex developmental disorder, and the hypotonia at birth might be misinterpreted as floppy infant syndrome. Patients with DBQD-2, presented with progressive scoliosis since early childhood. Unfortunately, no meticulous search for associated anomalies was performed and consequently no syndromic association has been identified.
The genetic cause of DBQD-1 and DBQD-2 has been identified in a number of patients, both are caused by biallelic pathogenic variants in either, the CANT1 gene for DBQD-1[4-6] or in the XYLT1 gene for DBQD-2 [7,8]. In addition to DBQD-1 and DBQD-2, a third type of the disease known as the Kim variant has also been defined with additional anomalies of hand bone [9-11]. This type is also inherited in an autosomal recessive manner, caused by variants in the CANT1 gene and has been included. CANT1 is located on (17q25.3) composed of three coding exons and encodes for a soluble calcium-activated nucleotidase 1 (CANT1) of 401 amino acids. CANT1 belongs to an apyrase family that preferentially hydrolyzes UDP followed by GDP and UTP. CANT1 was the first nucleotidase in the endochondral ossification process [5,12]. However, its exact function in human and bone formation is still unclear, its role in proteoglycan biogenesis and glycoprotein maturation has been recently determined [12]. Knock-out and knock-in experiments in mouse models, showed that connective tissue and skeletal phenotype were compatible with human DBQD1 and confirmed the essential role of CANT1 in proteoglycan biosynthesis process [12]. Up tuntil now and according to the Human Gene Mutation Database, about 27 pathogenic variants including missense, nonsense, frameshift, and the splicing site variants have been reported so far were associated with DBQD-1 [4]. XYLT1 (MIM 608124) is mapped to (16p12.3), consists of 12 exons and encodes for an enzyme of 959 amino acids called xylosyltransferase 1. XYLT1 was found to be essential for glycosaminoglycan (GAG) chains biosynthesis 8,13. Mouse model of XYLT1 showed reduced of GAG synthesis and skeletal phenotype recapitulate the human disease [14]. Around 17 pathogenic variants including frameshift, missense, splice site, and nonsense variants have so far been reported as associated with DBQD-2. On the other hand, a trinucleotide repeat (CGG) expansion in exon 1 of XYLT1 has recently been identified to be associated with hypermethylation and transcriptional repression of XYLT1 and responsible for Baratela-Scott syndrome (BSS; MIM: 300881)[15] . Moreover, a small deletion was also reported to be associated with severe short limb skeletal dysplasia in an infant but without typical clinical features of DBQD-II [16].
Here, we report eight patients (six girls and two boys) with an age range from 14-months to 14 years. Floppy infant syndrome was considered for children born with hypotonia. Scoliosis and kyphoscoliosis was the diagnostic entity for the second group of children. Both groups underwent a detailed and comprehensive clinical and radiographic documentation. Genetic analysis for five children manifested that the distinctive DBQD-1 was consistent with CANT1 mutations.
Ethical approval: The children enrolled in this study provided informed signed consent for their involvement in this research through their guardians, which were designated in accordance with the Helsinki declaration and approved by Ethics Committee of the (Ilizarov Scientific Research Institute, No.4(50)/13.12.2016, Kurgan, Russia)
All families are from certain geographical areas where they have lived for generations and were truly consanguineous.
Natural History
At birth hypotonia was the first and foremost clinical feature. Floppy infant syndrome was given to three children by the pediatricians. Floppiness combined with developmental retardation drove the pediatric neurologist to assume the diagnosis of congenital muscular dystrophy type Fukuyama (i.e. floppiness with brain involvement).
Pitfalls in Diagnosis
Cerebral CT scan did not reveal any form of cerebral malformation, no atrophy, no dilated ventricles, and no lissencephaly. In accordance with the results of the normal cerebral imaging and the relative muscular dystrophy, mutation in the FKTN gene was suggested by the geneticist. Though, neither CPK nor a muscle biopsy revealed any form of dystrophic changes. The genetic results of the FKTN gene mutation turned negative. The language, developmental skills and normal intelligence were the reasons to deter the diagnosis of Fukuyama syndrome. Though progressive cervical kyphosis and multiple joint contractures combined with floppiness, were the reasons to consider the diagnosis of arthrogryposis in connection with myopathic type of Ehlers-Danlos syndrome (Bethlem myopathy). Whole exome sequencing showed no collagenous mutation. Unfortunately, because of the wrong decisions made by the pediatricians, neurologists and the geneticists, the time elapsed between birth and referral to our department was too long and caused a negative outcome regarding the spinal and the articular corrections. Our fundamental strategy is based on comprehensive clinical and radiographic documentations. Clinically, all children referred to our departments exhibited pre and postnatal growth deficiency, revising their birth notes; marked decrease in birth length (-5SD to -7SD). Craniofacial examination characterized by round facies, long philtrum, saddle nose, hypertelorism, prominent eyes (in some) and flat nasal bridge with relative rhizomelia. All manifested a short narrow thorax associated with generalized ligamentous hyperlaxity and dislocations over the elbow and knee joints respectively. The necks are very short, and heads sit directly on the body. Musculo-skeletal examination showed generalized ligamentous hyperlaxity, multiple dislocations, over the elbows, inferior ulnar heads, and the knees.
Clinical documentations in our Departments
We subdivided our group of patients into two subgroups:
Desbuquois type 1(Phenotype)
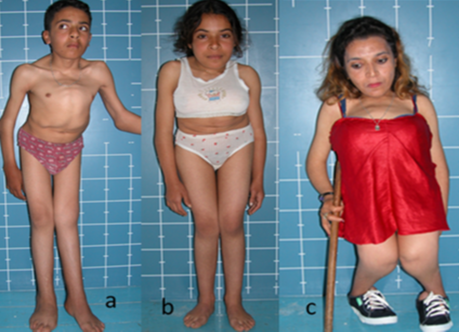
Marked floppiness associated with severe pre and postnatal growth deficiency of (-7SD). Five children have been categorized in this group (figure 1 a-e). The clinical phenotype is almost mimic each other. All manifested pre and postnatal growth retardation, prominent eyes, depressed nasal bridge, anteverted nares, mid-face hypoplasia, and round face. One girl manifested large frontal hemangioma (b). Three children manifested severe hypoplasia of the big toes (b,d,e).

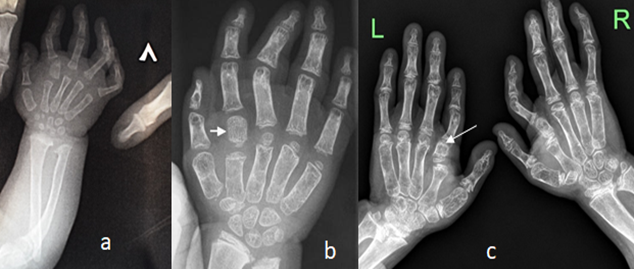
A.Hands: Hand radiograph of a 2-years-old-girl showed advanced bone age with supernumerary small carpal bones with evidence of accessory ossification bone (hyperphalangy) at the base of the second proximal phalanx (index finger) associated with multiple interphalangeal dislocations associated with a dislocated radial head (figure 2a). AP hands radiograph of a-5-years-old-girl showed advanced carpal ossification, short and broad first metacarpals, under-mineralization of the bones, note the hyperphalangy of the index finger (figure 2b).AP hand radiographs of a-7-years-old-girl showed advanced carpal ossification, short and broad first metacarpals, under-mineralization of the bones, note the hyperphalangy of the index finger (arrows) (figure 2c).
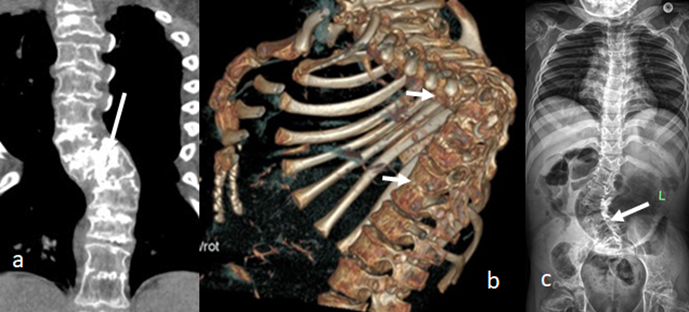
B.Cervical Spine: Sagittal cervical MRI in a -12-months-old-girl MRI showed massive impingement of the spinal cord at the level of C7 with early signs of myelopathy (figure 3a). 3D reconstruction CT scan of the skull and cervical spine in 8-months old boy showed marked skull bones dysplasia (note the cervical angular kyphosis at the level of C5-arrow (3,b). 3D reconstruction CT scan of the cranio-cervical junction in a 9-years-old-boy showed incomplete development of the posterior arch of the atlas (arrow head) and odontoid hypoplasia (arrow) (3,c).

C.Thoraco-Lumbar : Reconstruction coronal CT scan of a-6-year-old-boy showed progressive thoracic kyphoscoliosis because of butterfly vertebrae of most of the thoracic vertebral bodies (figure 4,a). 3D reconstruction CT scan of a-7-year-old-boy showed acute angular thoracic kyphosis of 90° Cobbs angle secondary to diffuse fusion of the upper thoracic and lower thoracic vertebrae (4,b). AP spine radiograph of a-7-year-old-girl showed progressive lumbar scoliosis in connection with butterfly vertebrae of L3-5, broad ribs associated with overwhelming osteoporosis (figure 4c).
D.Pelvis and lower Limbs: AP standing radiograph of a-2-year-old-girl showed epi-metaphyseal dysplasia and the proximal femora displayed the view of a monkey wrench appearance (arrow), bilateral dislocation of the knee joints (figure 5a) AP standing radiograph of the lower limbs of a -7-year-old-girl showed dysplasia of the capital femoral epiphyses resulted in the development of the monkey wrench or the Swedish key appearance of the proximal femora, hyper-lax and dislocated knees (arrow) associated with massive osteoporosis (figure 5, b) 3D reconstruction CT scan of the pelvis at the age of 8-months showed the Swedish key appearance in connection with pronounced lesser trochanter (arrow) associated with dysplastic capital femoral epiphyses (figure 5c). Hypoplasia of the big toe seen in all five patients of Desbuquois type 1 may be considered as an expansion of the skeletal phenotype.

Three siblings of (12, 16 and 20 year) respectively. A-12-year-old-male sibling showed normal facies but very similar skeletal malformation complex typical as seen in his female siblings (figure 6 a,b,c). The 20 year-old-girl manifested severe short stature (-8SD) and massive skeletal malformation complex (figure 6,c) .
This group of siblings showed a better developmental history. Needless to say, in late childhood they manifested progressive deterioration in their overall mobility. Early life ligamentous hyper laxity turned into massive and generalized axial and articular stiffness. Skeletal survey of this family showed: AP hand radiograph of a 12-year-male sibling showed disorganized ossification of the carpal bones with advanced bone age and supernumerary carpal bones. Interestingly, the typical hand changes in type 1 Desbuquois is not encountered in type 2 (figure 7a).

Lateral spine radiograph of the 16-year-old female sibling showed progressive thoracic kyphosis of Cobb’s angle of 80 ° (b). AP pelvis radiograph of the 16-year-old female sibling showed epi-metaphyseal dysplasia and the proximal femora displayed the view of a monkey wrench appearance (c). AP lower limbs radiograph of the 20-year-old-female sibling showed proximal femora displayed the view of a monkey wrench appearance, genu valgum and bilateral and symmetrical epi-metaphyseal dysplasia of the knees (d).
CANT1 gene sequencing in one family showed a homozygous pathogenic variant c.898C>T (p.R300H) lies in exon 4 of the CANT1 gene and alters a conserved residue in the protein. This variant is predicted to be damaging by insilico missense prediction tools (SIFT and Polyphen2). The identified variant has been reported in the dbSNP database with the identification number rs267606701 and is present in population databases such as genome Aggregation Database (gnomAD) as a rare variant with the highest allele frequency of (0.00001746). In the ClinVar database, the clinical significance of this variant has been reported as ‘pathogenic’. The identified variant was previously reported in two Turkish and one Iranian patients with the relevant phenotype consistent with the features of DBQD-1 [5]. Based on ACMG guidelines, this variant was classified as ‘pathogenic/ Likely pathogenic’ results in DBQD-1. Parents were heterozygote for this mutation.
Desbuquois syndrome is reminiscent of Larsen syndrome Maroteaux et al. In that there is joint laxity with multiple dislocations [17]. The eyes are prominent, the nasal bridge tends to be flat, with potentially marked micrognathia, but the bone changes are quite different, and the radiological changes are distinctive in Desbuquois syndrome: there are supernumerary phalanges, characteristically situated between the metacarpal and proximal phalanx of the index finger, osteoporosis, a short narrow thorax, metaphyseal enlargement and platyspondyly. Furthermore, the spurs of the lesser trochanters are constant signs. Maroteaux et al., (1986) suggests that the prominence of the lesser trochanter at the hip is characteristic. Ossification in the carpal centers may be advanced, whereas the epiphyses of the long bones can be retarded. Generalized ligamentous hyperlaxity, joint dislocations, progressive scoliosis/kyphoscoliosis, monkey wrench or Swedish key appearance of the proximal femora and advanced carpotarsal ossification are characteristic and diagnostic signs.
Al Kaissi et al (2005) described a 3-generation Tunisian family with a novel type resembling Desbuquois syndrome [18]. Al Kaissi et al (2009;2013) described the diagnosis and the orthopedic Management of Austrian Patients with Desbuquois type 1[19-21].
Bernd et al., (1990) pointed out the similarity of the hand changes to those seen in Catel-Manzke syndrome, described a detailed report of an infant first briefly reported by Catel in 1961 [22,23]. The key features are micrognathia, cleft palate, glossoptosis and an accessory (usually triangular) bone at the base of the index finger. The latter was also found in our patients, but the rest of the clinical and radiological criteria were different. Clinically, the children with Catel-Manzke syndrome, have the appearance of severe Pierre Robin associated with a short, flexed and ulnar-deviated index finger.
Classically, Desbuquois syndrome presented in two types in accordance with the presence or absence of hyperphalangy of the index finger. In DBQD-1, hyperphalangy is a characteristic radiological feature (an extra ossification center between the proximal phalangeal base and distal end of the metacarpal) which is categorized as DBQD-1. DBQD-2 is the same without hyperphalangy, though characterized by accelerated carpo-tarsal ossification.
Patients with Desbuquois dysplasia manifest marked disease-course variability in accordance with the type. For instance, type I (hyperphalangy) as infants were categorized with the distinctive category of floppy infant syndrome. DBQD-1 seems to be a complex developmental disorder sharing clinical features with other syndromic entities such as congenital myopathy. The clinical reports of the pediatricians were almost uniform and telling similar stories. They described these infants as severely hypotonic and fit within the category of muscular disease of arthrogryposis. In that the infants seem to lie right in mattress instead of on it, the limbs stayed wherever they are put, the arms lie out on either side of the head and the legs take up the characteristic (frog) position of abduction and external rotation at the hips. The proximal muscles look extremely hypotonic but not paralyzed, and there were minimal movements of the hands and feet. The tendon reflexes are brisk but not absent, no fasciculation of the tongue has been elicited. Breathing noticeably shallow and diaphragmatic. When picked up, the infant´s head falls back with reduced tone in the shoulder girdle, which leads these infants to slip through the hands of the examining pediatrician. In addition, when consanguinity combines with the clinical picture of floppiness, the suspicion among pediatricians, pediatric neurologists and geneticists of an autosomal recessive disorder of congenital muscular dystrophy becomes strong. The severe hypotonia associated with some contractures and the psychosomatic delay were the main parameters to include this group of infants within the congenital muscular dystrophy type Fukuyama (i.e. floppiness with brain involvement). Misdiagnoses continue to show along with insufficient history taking and a lack of clinical skills in understanding what lies behind the infant /child´s true clinical picture. The advancements in pediatrics have necessitated the addition of dysmorphology as new clinical material throughout the training courses of pediatricians and pediatric neurologists. The basic approach to save time, efforts and to avoid the adverse psychological impact on parents and families, requires better comprehension in syndromic associations. Invisible, though essential clinical criteria for clinical diagnosis are mostly overlooked. Sadly speaking, the general notion among pediatricians in that syndromes are rare, caused immense damage to and choose other diagnostic paradigm such as idiopathic, arthrogrypotic and so forth.
Authors Contributions
AAK, SV and FBC (Clinical and Radiographic documentation, and writing the MS). SBC,VK, PO, SR and FG surgical intervention, LBJ, MS, and SGK performed screening for lysosomal storage disorders and whole exome sequencing. All authors approved the final version.
Acknowledgments
We wish to thank Ms. Katharina Sigl, head of the musculo-skeletal group (0rdensklinikum) Linz, Austria for her help in facilitating the required investigations.
Conflict of interest
The authors declare that they have no competing interest
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
