
Case Report | DOI: https://doi.org/10.31579/2578-8949/069
1 Second year resident of Internal Medicine at Hospital Ángeles Metropolitano.
2 Dermatologist and Dermato-Oncologist Private Practice
3 First year resident of Dermatology at Dermatological Center "Dr. Ladislao de la Pascua
4 Department of Allergy and Clinical Immunology Internal Medicine, Hospital Ángeles Mocel Gobernador Gregorio V. Gelati St. Number 29 Col. San Miguel Chapultepec I Secc, Miguel Hidalgo, 11850 Mexico City.
*Corresponding Author: Valente Maldonado MD Msc, Department of Allergy and Clinical Immunology Internal Medicine, Department of Allergy and Clinical Immunology Internal Medicine, Hospital Ángeles Mocel Calle Gobernador Gregorio V. Gelati Number 29 Col. San Miguel Chapultepec I
Citation: G F Ramos, E L G Sánchez, V Á Rivero, V Maldonado. (2020) Dermatomiositis anti-mda5 positive Report of a case and literature review. Journal of Dermatology and Dermatitis.5 (2); Doi:10.31579/2578-8949/069
Copyright: © 2020 Valente Maldonado, This is an open-access article distributed under the terms of The Creative Commons. Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 07 July 2020 | Accepted: 07 August 2020 | Published: 11 August 2020
Keywords: dermatomyositis; clinically amyopathic dermatomyositis; anti-mda-5 antibodies; autoimmune; muscle weakness; skin manifestations
Dermatomyositis (DM) is an autoimmune connective tissue disorder with characteristic skin changes and involvement of different organs and systems, such as muscle, cardiovascular, respiratory and gastrointestinal systems.
This condition is characterized by the presence of a myopathic syndrome with bilateral and symmetric weakness predominantly of extensor, proximal muscle groups, respiratory muscles and esophageal involvement, accompanied by characteristic skin disorders.
The worldwide incidence is reported at 1 to 6 cases per million inhabitants; predominates in women, with 2.5:1 ratio; and has a bimodal distribution with two peaks, one from 5 to 14 years old and another from 45 to 64 years. The amyopathic form is present in 2 to 18% of all cases of dermatomyositis.
Diagnosis is supported by alteration in muscle enzymes, as well as characteristic findings in magnetic resonance imaging (MRI), electromyography (EMG) and autoantibodies. The definitive diagnosis is determined by the histopathological study of both, skin biopsy and affected muscles.
The purpose of this case is to review the diagnostic approach from a dermatological point of view, where characteristic lesions gave way to a thorough physical examination looking for signs of a myopathic syndrome; and where despite the elevation of muscle enzymes, findings in electromyography and MRI compatible with myositis, the muscle biopsy was negative. It was the punch biopsy that corroborated the diagnosis, even before the report of anti-MDA-5 antibodies (melanoma differentiation associated Gen 5) that has been linked to a characteristic phenotype of DM that has a higher prevalence of amyopathic disease and a worse prognosis.
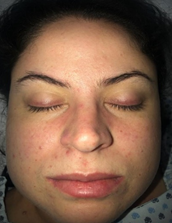
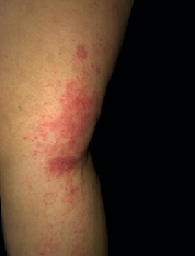
A 27-year-old woman with no relevant medical history. That after prolonged sun exposure and consumption of hallucinogenic mushrooms, she presented a dermatosis located at the V of the neckline characterized by erythematous-violet spots of specific edges. Besides, in her eyelids, she had heliotrope rash. (Image 1) As well as oral ulcers at the cheek level, she also had erythematopapular plaques located in metacarpophalangeal joints (Gottron’s sign and Gottron’s papules) (image 2). Furthermore, she had plaques of lower-border erythematous papular in the limbs (Image 3). Subsequently, she had pain and weakness in the lower extremities, making it difficult to change the position from sitting to standing and impossibility of ambulation, affecting the proximal muscle groups of the upper extremities, causing bedridden prostration.

To the physical exam, she showed a weakness in all her extremities with muscle strength 3/5 on the Daniels scale in the lower extremities, and 2/5 in the upper extremities, with a preserved sensitivity.
In tests, the most relevant data was Creatinephosphokinase up to 22 084 (U / L).
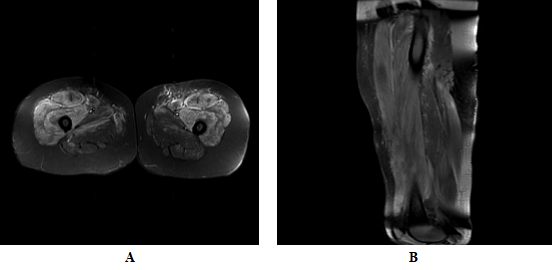
On the other hand, comparative MRI of quadriceps showed generalized intramuscular edema with more evident inflammatory changes in muscle groups of the anterior compartment (quadriceps femoris), with a slight right predominance, in addition to inter and perifascial edema (Images 4A and 4B), findings that affect the myositis process.
Data of myopathic alteration in the four extremities with abundant data of membrane instability.
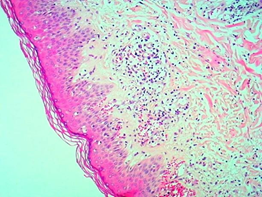
Skin biopsy of the right leg and arm performed with a 5 mm punch shows orthokeratotic hyperkeratosis, follicular plugs, flattening of the epidermis with vacuolization of basal cells, accentuated edema in the dermis, telangiectatic dilation of the superficial plexus with lymphocytic infiltrate, punctuated with occasional eosinophils. Histological changes were compatible with Dermatomyositis. (Images 5, 6 and 7)


We decided to run an antibody panel test that was Anti-MDA5 positive.

Dermatomyositis is an autoimmune disorder, with characteristic changes in the skin and involvement of many organs, including muscles, blood vessels, joints, gastrointestinal and respiratory systems.5 at the lung level, there is a strong association with malignancies.[6]
In DM inflammation of the striated, bilateral, symmetrical, extensor and proximal musculature, accompanied by characteristic skin disorders are typical. [2]
Among the different varieties of DM, the gene 5 associated with melanoma differentiation (MDA5) has been identified as a specific autoantigen of DM that seems to be aimed at patients with DM and mild or absent muscle inflammation and with an increased risk of disease interstitial lung.[27]
Fiorentino et al. describe a particular phenotype in patients with anti-MDA5 positivity, which consists of a characteristic skin pattern, with the presence of ulcerations in the oral cavity, on nail borders, elbows and over Gottron's papules; also, these patients had a higher risk of joint affection, panniculitis, and alopecia, in the absence of significant myositis, and a higher risk of developing interstitial lung disease. [27.29]
The role of dermatologists is crucial, due to the difficulty in recognizing the disease in the absence of muscular manifestations, which often leads to delays in treatment, which can influence the quality of life and prognosis of the patient. [10]
DM predominates in women, with a 2.5:1 ratio.2.3 It has a bimodal distribution of presentation, with two peaks, from 5 to 14 years old and from 45 to 64 years old.1-5 A cumulative incidence of Dermatomyositis of 1 to 6 cases per million inhabitants has been described.[2, 3] While the annual incidence is 2 to 10 cases per million and the prevalence is 5 to 8 cases per 100,000 inhabitants.2 It is worth mentioning that the incidence is increasing with 1 case per 100,000 people per year.10
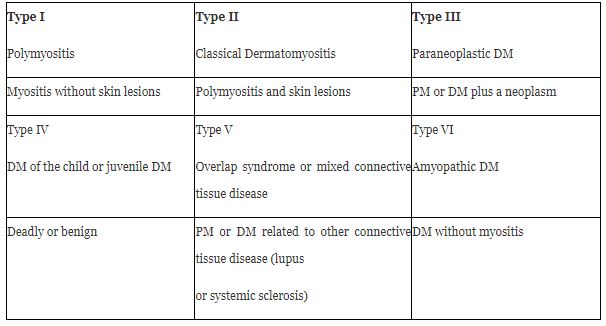
Among idiopathic inflammatory myopathies is DM.1 There is a subset of patients with DM (2-18%)2 who have a predominant phenotype of skin condition and are classified as clinically amyopathic DM (CADM) and in turn, these are subclassified into amyopathic and hypomyopathic.[2,4,9,10] Although both subtypes do not have clinical evidence of muscle involvement, there is subclinica evidence demonstrated in the laboratory, electrophysiological, or radiological evaluations of the hypomyopathic variant.[9,10,31,33] (Table 1)
The specific cause of DM is unknown. 2.3 has been associated with factors environmental, infectious, viral, endocrine, and genetic, triggering autoimmune processes; in genetically susceptible individuals.
There are even cases related to the use of certain medications such as hydroxyurea, nonsteroidal anti-inflammatory drugs, quinidine, d-penicillamine, and tumor necrosis factor antagonists. [4.5]
Similarly, 15 and 25% present an underlying neoplastic process.2-5 Among the most frequently related neoplasms, they are found in the lung, breast, ovary, stomach, lymph nodes, spleen, bone marrow, colon and rectum, uterus, prostate, and skin. [2, 4, 7]
Among the evidence that supports the immunological origin of DM, it is found that in 60 to 80% of patients, nuclear and cytoplasmic autoantibodies specific or related to myositis are detected.2
It is possible that genetics, probably plays an essential role in the risk of developing DM. Since specific alleles of human leukocyte antigen (HLA) are risk factors for the development of DM. [1.5]
In the specific case of DM associated with anti-MDA 5, it is essential to mention that MDA5 recognizes viral nucleic acids and triggers the production of type 1 IFN, thereby suppressing viral replication.29 It has been shown that mutations in the IFIH1 gene encoding MDA5 lead to interferon driven auto inflammatory diseases.30
Cutaneous manifestations
The heliotrope rash and Gottron’s papules are pathognomonics signs of DM [4, 5, 10]
The dermatological features are the presence of patches and plates that have a pink color to be erythematous-violaceous photosensitive, which appears symmetrically over bony prominences as knees, elbows, interphalangeal joints, and exposed areas. [4, 5, 10]The rash's natural history is chronic and often progressive, ultimately resulting in poikiloderma, depigmentation, atrophy, and telangiectasias. [1, 2]The skin condition occurs in 30 to 40% of adult patients with classic DM and 95% of the cases of juvenile DM. [5] These usually precede to the appearance of Myositis between 3 and 6 months and one year in 30-50% of patients; 4,5,10 while only 10% of patients present muscle symptoms before the development of the appearance of skin lesions [10]
Skin changes in DM are distributed to prototypical regions in the body, such as1
It is violet erythema, with slight edema and scaling that appears on the eyelids, forehead, malar regions, and back of the nose; so named because of its similarity to the violet petals of the Heliotropium peruvianum flower6 and occurs in up to 74% of patients.2-5
confluent purplish erythema is located on the back of the neck, upper back, and shoulders, extending to the upper arms.1-5. [10]
it is confluent violet erythema that acquires a poikiloderma appearance in the sun-exposed areas of the anterior and inferior neck and the anterior thorax. 1-5. [10]
Purplish to pinkish papules on the interphalangeal and metacarpophalangeal joints, present in up to 80% of patients. 1-5. [10]
The presence of painful papules on the palms represents a unique characteristic of the presence of antibodies to anti-MDA5.29
It is purplish erythema that appears symmetrically on the interphalangeal joints, the olecranon processes, the patella, and the medial malleoli. [1, 3, 4, 5, 10]
This is the name given to purplish macular erythema and poikiloderma on the lateral hips and lateral thighs. [1, 10, 2, 4, 5]
Recently, the "sleeve sign" has been described, which consists of purplish macular erythema restricted to the lateral aspects of the upper arms; this location is compatible with the sleeves' contour. [5]
Other characteristic findings include the so-called "mechanical" hands; this consists of hyperkeratosis and fissures along with the thumb, second and third fingers.1.5
The scalp is frequently affected in DM, and characteristic changes include erythematous squamous plaques that resemble severe seborrheic dermatitis or psoriasis.5,10 Often, the scalp rash is accompanied by generalized non-scarring alopecia. [5.10]
Other manifestations include the appearance of cutaneous calcinosis (20%), which can affect the skin (calcinosis cutis), joints (circumscribed calcinosis), and muscular fascia (calcinosis Universalis) that can cause disability. [2,3]
Myositis usually presents as symmetrical proximal muscle weakness.1-4,9 Patients often report weakness in the extensor muscles surrounding the shoulder and pelvic girdle.1-4,9 Weakness of the quadriceps and gluteal muscles can manifest as difficulty in getting up from a chair and climbing stairs, which causes the characteristic so-called anserine gait.1-3 Patients may report weakness in the shoulders and upper extremities as difficulty washing their hair or reaching for objects in the upper cabinets.1-3 In 30% of patients, the main discomfort is muscle pain. [1]
The involvement of the respiratory muscles of the chest wall or diaphragm can also cause respiratory distress. [1,3] The involvement of the cricoarytenoid muscle can occur in up to 40% of patients and presents as dysphonia.22 Dysphagia can occur in 20% to 50% of cases due to the pharyngeal musculature weakness. [3.4]
The clinical manifestations of small vessel cutaneous vasculitis vary and may include petechiae, palpable purpura, urticarial lesions, livedo reticularis, and skin or oral ulceration in up to 30% of patients.5 It often affects the skin over the extensor articular surfaces, although it can be found anywhere.5,6 Skin ulceration in DM guarantees the presence of anti- MDA5 antibodies or malignancy. [1,5,6,29] This tends to happen on Gottron's papules or in areas where the Gottron´s sign is, nail edges, and pulp finger region. [1, 29]
Another common feature is the involvement of the proximal nail fold; edema, capillary dilation, and cuticular hemorrhage can be observed, These findings may represent disease activity.2-6 The Samitz sign refers to dystrophic and irregular cuticles. [5]
A symmetrical purplish patch can be seen in the midline of the hard palate; constant location in the center of the hard palate can aid in the diagnosis of DM.1 Other findings of the oral mucosa include gingival telangiectasias, as well as more classical lichen planus-like lesions around the gingiva or oral mucosa. Moreover, painful sores on gums, tongue, and palate are associated with the positivity of anti-MDA-5.29
Interstitial lung disease (ILD) is the most common pulmonary manifestation in DM, affecting between 15% and 50% of patients, and it is one of the leading causes of morbidity and mortality.4,10,20 Large case series have suggested that more than 75% to 86% of patients who have anti-synthase antibodies will develop ILD . DM patients with anti-MDA5 antibodies have a markedly increased risk of developing ILD with estimates between 50% and 100%. 5,10,16,20,25 The presence of skin ulcers in the context of positivity for anti-MDA5 antibodies represent a strong predictor for the development of ILD.29 Patients should be examined regularly for lung symptoms, given the prevalence of interstitial lung disease.[10]
A rare gastrointestinal condition is reported in 4% of young patients with DM, and its presence is associated with more severe disease.2 However, kroner et al. report that more than 50% of patients have gastrointestinal involvement.22These symptoms include persistent and progressive abdominal pain, nonspecific symptoms such as diarrhea, vomiting, constipation, dysphagia, heartburn, nasal regurgitation, and hoarseness due to weakness of the proximal esophageal skeletal muscles.2,22 More serious ones include ulceration and perforation, which can be life-threatening. [1.2]
Cardiac involvement in DM is increasingly recognized as an essential clinical feature and is usually subclinical; 1, 2 about 50% of patients are asymptomatic.25 Cardiac manifestations include arrhythmias, congestive heart failure, myocarditis, pericarditis, angina, and fibrosis. [25]
A variety of DM has been described, in which the individuals present the characteristic cutaneous manifestations, however in the absence of significant muscular affection, that is to say, absence of muscular weakness or of the elevation of muscular enzymes, known as clinically amyopathic Dermatomyositis (CADM) [31] (formerly known as DM without Myositis) [2]

In 1975, Bohan and Peter established the diagnostic criteria for dermatomyositis, in force to this day. [33] (Table 3)
Diagnostic studies include biopsy of muscles with weakness, a biopsy of the affected skin, electromyography, and measurement of muscle enzymes. [4, 6]
Sontheimer, proposes the formal skin criteria to make the diagnosis of DM. Three major criteria include the heliotrope rash, which is considered pathognomonic, Gottron's papules, Gottron's sign, and 14 additional criteria.[34] (table 4)
Sontheimer suggested that the diagnosis of CADM could be made if two major criteria or a major criterion were present and the presence of two minor criteria in addition to the biopsy of at least one region showing histopathological changes consistent with DM. However, these criteria are not validated. [34]
For the diagnosis of CADM, a confirmatory skin biopsy is required, in addition to the fact that the patient had not been under immunosuppressive therapy for two consecutive months or more, within the first six months of the onset of skin disease, since such therapy could mask or delay the development of myositis.[31]
Myositis is not a requirement for diagnosis. However, the evidence supporting the presence of inflammatory myopathy is useful when a patient has a suspicious rash. 2-6


Elevation of muscle enzymes should be the next step in defining the diagnosis.6,9 At the onset of Myositis, serum muscle enzymes (creatine kinase, aldolase, lactate dehydrogenase [LDH], aspartate aminotransferase [AST]) and alanine aminotransferase [ALT]) are reasonably sensitive biologic markers of muscle inflammation1-4,6,9; the creatinine phosphokinase ( CPK ) is the most sensitive and specific.2 However, in the middle or late in the course of myositis, its sensitivity decreases.1 It has been observed that in cases of paraneoplastic dermatomyositis (type 3), the enzymes are usually normal.3 Measurement of 24-hour urine creatine concentration is an early and sensitive indicator of muscle injury and a better indicator of activity than serum creatine kinase concentration. The test is especially useful when the serum creatine level is average. [4]
Most DM patients only have an MSA, and only about 20% of DM patients have a known MSA.10 However, other literature reports its presence in up to 60% of patients. [9]
These MSAs can provide essential information to establish the diagnosis of DM in complex cases. [4, 6, 9, 10] It is critical to highlight that the MSAs are currently used to phenotype and stratify patients with DM rather than make the diagnosis; however, they can be helpful to confirm it when they are present. [9, 10]
Its presence varies between 2 to 38%. The patients present cutaneous and muscular findings; they have a lower incidence of ILD and malignancy.9, 10 In general, patients have a favorable prognosis and respond well to therapy. [9, 10]
Positive from 1 to 10%. These patients tend to have classic skin findings, myositis and dysphagia. [9, 10]
These include anti-Jo-1, anti-OJ, anti-EJ, anti-KS, anti-Zo, anti-Ha / YRS, anti-PL-12, and anti-PL-1. They have been associated with the anti-synthetase syndrome. [1, 4, 6, 9, 10, 11]
The DM anti-MDA5 exhibits unique clinical features including mucocutaneous ulceration, palmar papules, scarless alopecia, panniculitis, arthritis, and interstitial lung disease. [29] Its positivity is related to greater disease progression and higher mortality.26It has been observed that patients with MDA-5 positive DM have a higher prevalence of amyopathic disease. [10, 11, 16] Liubing Li et al, reported that the presence of anti-MDA5 antibodies was associated with CADM and not with PM. [28]
There is a well-established association of malignancy in patients with anti-TIF-1γ antibodies; the risk may be influenced by various factors, such as male sex, advanced age, and smoking. [8, 9, 10]
Further studies are needed to determine whether these clinical factors and antibodies can be used to stratify patients with DM based on their risk of malignancy, but currently, detection of malignancy is recommended for all adult patients diagnosed with DM. [10]
Electromyography or magnetic resonance imaging can be used in situations where clinical suspicion remains high for myopathy, but muscle enzymes are normal. [4,9] Myositis can be detected by electromyography in 70%-90% of DM patients with active muscular disease, and the sensitivity to detect myositis decreases over time.9 The classic myositis triad includes short-duration, small-amplitude polyphasic motor unit potentials, fibrillation, acute positive waves, and complex repetitive discharges. [2, 3, 9] These findings reflect active muscle inflammation and can be seen in other inflammatory myopathies such as polymyositis.1-3. [9]
Magnetic resonance imaging provides a detailed view of the muscular anatomy, which allows assessing the location and discrimination of the type of pathological process (edema, inflammation, fibrosis, calcifications, or atrophy). [4, 6, 9]
MRI can also be used to direct the site of a diagnostic muscle biopsy or can be used to assess the clinical response to treatment. [1, 4, 9]
The association between dermatomyositis and malignancy is well established.8 DM is considered a paraneoplastic phenomenon, and cancer may precede or follow the development of clinical signs of DM. [5, 7, 10]
Compared to the general population, the incidence of cancer is estimated to be 5 to 7 times higher in patients with DM.8 This association seems to correspond more closely with dermatological manifestations since it occurs more frequently in patients with CADM.4 Detection of malignancy is critical because the treatment of associated cancer can reduce the severity of the disease and, sometimes, its remission. [4.7] since 10% to 20% of patients with DM have an associated malignancy, extensive investigation is warranted at initial diagnosis.4.7 currently, there are no agreed guidelines on screening. [5, 7] A general screening approach may include pelvic ultrasound and mammography in women, prostate examination and testicular ultrasound in men, fecal occult blood, colonoscopy, and computed tomography of the chest, abdomen, and pelvis in both sexes to detect any hidden malignancy. [5, 7].
The independent risk factors for presenting an underlying neoplasm are age> 52 years at diagnosis, abrupt and rapid onset of skin lesions and muscle weakness, presence of necrotic lesions and periungual erythema, and low C4 levels. [2, 4, 8] Bowerman et al, also describe that autoantibodies against TIF-1 γ and nuclear matrix protein 2 (NXP-2) have been associated with an increased risk of presenting malignancy. [8] Kundrick et al, highlight in their study that younger adult patients also have a considerably higher risk of cancer, suggesting that a screening approach only for the older population increases the risk of losing a significant proportion of hidden malignant tumors in young patients with DM. [7]
The development of evidence-based guidelines is needed to optimize malignancy detection in adult patients with DM of all ages. [7]
The gold standard for detection is high resolution computed tomography (HRCT). It is recommended to be performed every 3-6 months during the first year of diagnosis. [29]
Biopsies of affected skin in DM classically show cell-poor interface dermatitis, increased dermal mucin, perivascular lymphocytic infiltrate, CD3 + T cell infiltration, plasmacytoid dendritic cells, macrophages, as well as B cells and vascular ectasia. [4, 6, 10] Mild hyperkeratosis and atrophy areas are observed in the epidermis, as well as liquefaction of the basal (interface damage). [2, 4, 10]Edema of the connective tissue, fibrinoid collagen necrosis, and vasodilation with perivascular lymphocytic infiltrates can be observed in the dermis. [2] It has been suggested that DM is characterized by an increased presence of plasmacytoid dendritic cells mainly of epidermal location, whereas in cutaneous lupus, they are mainly found in the dermis. [1] Since these findings are not specific to DM, DM lesions should be identified clinically, especially with their distribution. [9]
In patients with characteristic skin lesions and muscle weakness, a muscle biopsy is rarely necessary to establish DM diagnosis. [1]
The results depend on the technique when taking the sample, since if the muscle fibers are allowed to contract after cutting them, the study may not be conclusive when the muscular alterations are not observed.3 the sample should be taken from a weak muscle, usually from a proximal muscle group, such as the biceps or quadriceps. [4]
The findings depend on the degree of muscle attack and the early or late stage in which the disease is found. [3]
Typical findings of muscle histopathology in DM include perifascicular atrophy, regenerated and degenerated myofibers, complex deposition of membrane attack on endomysial capillary walls, inflammation of endothelial cells, and capillary necrosis.3 Alteration of chromophilia and perivascular lymphocytic infiltrates, as well as a loss of normal striation due to the presence of fibrinoid substance, are also observed. [2.3]
Muscle biopsy can give false-negative results, both for immunosuppressive drugs and for the unpredictable nature of inflammation. [1]
The course of skin disease tends to be chronic and long.10 A prospective cohort of 74 patients with DM who received various systemic regimens found that only 38% had achieved remission of the skin disease in a 3-year follow-up period. [10]
There is no effective medicine in all cases.3 Treatment of DM first requires evaluating the potentially affected organs (skin, muscles, and lungs). [1] with regard to skin disease, specifically, the goal of treatment is to gain control of skin disease using the safest therapeutic combination. [10]
First-line therapy should include aggressive photoprotection, antipruritic agents, and topical anti-inflammatory medications (corticosteroids and calcineurin inhibitors). In the vast majority of patients with DM, these therapies should be used as adjuvant therapies to systemic agents, given the refractory nature of cutaneous manifestations. [4, 5, 10, 11]
Photoprotection: Since it has been proven that ultraviolet light can act as a trigger and exacerbator for skin lesions, the use of broad-spectrum sunscreen (SPF> 50) is recommended.6,10 This should be used daily and reapplied every 2 hours, in addition to avoiding exposure to prolonged sunlight.[4,6,10]
Handling pruritus: Sometimes, the pruritus in DM may seriously affect the quality of life of patients, this can be handled with emollients to reduce xerosis in combination with oral antihistamines, and other agents with antipruritic property as amitriptyline and gabapentin. [10]
Topical corticosteroids: can have a palliative effect on skin disease by reducing erythema, itching, and playing an adjunctive role to systemic agents.2,4,10 However, they are unlikely to control skin symptoms, except in milder cases.1 Class I or II topical corticosteroids are used on the thick skin of the elbows, knees, and hyperkeratosis on the hands and plastic-wrapped occlusion can be added at night to increase potency even further,10 while Lower potency (groups VI and VII) can be used on thinner and atrophy-prone areas, such as the face. [10]
Topical calcineurin inhibitors: These include tacrolimus 0.1% or pimecrolimus 1%; these may have moderate efficacy in some cases of cutaneous DM.2-5,10They offer the benefit of being used safely on the face without worrying about atrophy or hypopigmentation. [5, 10]
Systemic corticosteroids: Oral corticosteroids (OCE) are the first choice treatment. [4, 6, 9, 11, 23] OCEs are undesirable agents as monotherapy for cutaneous DM because they generally elicit partial responses and are associated with long-term side effects.1 However, they are the first-line therapy in the treatment of myositis. [10, 23]The American Academy of Dermatology recommends starting doses of oral prednisone (PSL) of 0.5 to 1.5 mg/kg / day. 9,10,18,23
The timing of glucocorticoid reduction should be decided based on the activity of muscle disease and extra muscular implications. Treatment with initial doses of glucocorticoids is generally continued for 2 to 4 weeks.9 Retrospective studies mention that the most notable improvement in DM occurs after 6 to 12 months of aggressive steroid treatment. [11,14,15] Intravenous pulse therapy with methylprednisolone (MP) may be necessary in cases of severe disease with multiple organ involvement, an initial dose of 1 g / day is recommended for three consecutive days followed by high doses of oral corticosteroids for a prolonged period. [18,23] It has been observed that MP pulse therapy can shorten the time to achieve remission. [9]
The addition of corticosteroid-sparing agents can improve the control of myositis and extracutaneous manifestations.23 they also help reduce the risk of presenting side effects due to the use of steroids for a long time. [4, 5, 11, 14, 15]
Findlay et al, suggest adding a second-line immunosuppressant to the OCE regimen, if there is no improvement after three to six months, or if an outbreak occurs when you stop taking steroids.19
Antimalarials: Antimalarials are generally considered first-line agents for cutaneous manifestations. [2, 5] Their exact mechanism of action is unknown. However, it is well known that they possess anti-inflammatory and photoprotective activity. [9,10] Hydroxychloroquine sulfate (200 to 400 mg/day) and adding quinacrine (100 mg twice daily) is recommended if the response is slow.4,10 Response to antimalarials is generally not evident until 6-8 weeks after onset. [9, 10]These agents have no activity in muscle disease. [4, 9, 10]
Methotrexate (MTX): it is an antimetabolite with antiproliferative and anti-inflammatory properties.9 Methotrexate is a first-line adjuvant therapy in patients who do not respond to steroids;4 is effective in significantly reducing the severity of skin disease in 50% to 100% of patients with DM at low doses of 5-15 mg weekly. [1, 4, 5] Several retrospective studies reported that concomitant treatment of MTX and PSL was effective in patients with PM / DM who did not respond to prednisone treatment. [9] Importantly, it is useful for both cutaneous and muscular diseases, making it an excellent corticosteroid-sparing agent.10In a multicenter randomized study, a better response was demonstrated when combining MTX or cyclosporine with prednisone early compared with prednisone alone, in patients with recent- onset juvenile DM; a shorter time was observed in reaching remission, and it also decreased the use of OCE. [24]
Mycophenolate Mofetil (MMF): is a selective lymphocyte immunosuppressive agent that inhibits inosine monophosphate dehydrogenase, an enzyme necessary for de novo purine synthesis.9,10 MMF has been shown to be effective at doses of 2 to 3 g / day to reduce skin and muscle disease severity.4 MMF is a useful agent for skin disease, myositis, and associated interstitial lung disease; It is the preferred first-line agent as it has been shown to allow improvement in lung function and to have a corticosteroid-sparing effect. [18]
Some authors prefer to use MMF agent saving of initial corticosteroid in patients with DM anti-MDA5. MMF has been shown to be effective in treating anti-MDA5 DM in case reports and small series. [29]
Morganrotht et al, in a series of cases of patients with DM and ILD who received MMF coupled to PSL, showed complete remission of the interstitial opacities observed on CT, normalized their lung function tests, improved dyspnea and allowed a reduction in use of prednisone at one year of follow-up. [17]
Intravenous immunoglobulin (IVIg): IVIg is probably the most effective agent for cutaneous DM, with 70% to 80% almost complete or complete response.2,4,5 It works by neutralizing autoantibodies, downregulating proinflammatory cytosines, complement binding, and decreasing the formation and deposition of the membrane attack complex.9 IVIg is valid for both refractory skin disease and myositis. [9,10] The standard dosage regimen is 2 g / kg/month divided into 3 to 5 days.5 Galimberti et al., observed in their retrospective study in 42 patients with refractory cutaneous disease that the addition of IVIg showed an improvement in 83% of the patients and decreased the use of OCE. [21]
Cyclosporine: The addition of cyclosporine was shown to play an essential role in the treatment of ILD related to Dermatomyositis, which is the most frequent cause of death in patients with DM.18,20,23 Jin Go et al, observed that early treatment with cyclosporine improved survival and slowed ILD progression in a group of 47 patients with DM and pulmonary manifestations.20 Ruberto et al., in their clinical trial that included 139 patients with recent onset juvenile DM and without prior treatment, demonstrated that initial therapy with prednisone and cyclosporine or methotrexate was more effective than prednisone alone. [24]
Rituximab: It is an anti-CD20 monoclonal antibody that has been shown to be effective in treating dermatomyositis. [5.9, 11] Several studies suggest that may be beneficial in cases of refractory myositis. [11, 23] especially those with anti-Jo-1 and anti-Mi-2 positive autoantibodies. 1,2 Huang et al, in their case series, included 21 patients with anti-MDA-5 positive DM and reported that the use of cyclophosphamide or MMF combined with a calcineurin inhibitor or rituximab could be effective in these patients. [32]
Azathioprine (AZA): recommended as one of the first-line immunosuppressive agents in refractory disease.18 Concomitant treatment of MTX and AZA is a therapeutic option in refractory patients.9 Studies report that patients with PM treated with AZA and PSL required lower PSL doses for maintenance therapy and had better long-term physical functions than those treated with PSL alone. [9]
Abatacept: is a fully human fusion protein of CTLA-4 and the Fc portion of human IgG1.9,11 Prednisone doses have been shown to be successfully reduced after treatment with RTX or abatacept in most patients with refractory PM / DM. [9]
New treatment options such as Toll receptor inhibitors and stem cell transplantation are actively being investigated, although clinical trials are still lacking. [5]
Physical therapy in patients with DM and PM can stabilize disease progression, improve muscle strength and endurance.23 Physical therapy is very important in the management of DM to prevent atrophy and contractions. [4] An aerobic workout, including cycling and resistance exercises for 12 weeks, three times a week, showed improvement in muscle strength and was well tolerated. [2, 3]
Cutaneous dermatomyositis affects dramatically the patient's quality of life, because they involve exposed and visible areas of the skin, and itching can sometimes be disabling. [12] More significant skin activity is correlated with poorer quality of life. [12]
Werth et al. developed a validated tool used to measure the primary outcome in dermatomyositis clinical trials, the severity index and area of affection in DM, CDASI for its initials in English. [13]
There is a poor prognosis when muscle weakness has existed for more than four months before diagnosis, those with dysphagia, lung disease, malignancy, and DM patients with no creatine kinase elevation. [4]
Mortality is 25%, other authors mention a 5-year survival, only 50%25, but the disease can be controlled with proper treatment, which improves the survival in 85% of cases. [2, 3] The prognosis is better in amyopathic forms since it generally does not progress to myopathy, although it can be associated with malignant disease.2 Recurrence of edermatomyositis may indicate the appearance of a second primary malignancy or recurrent cancer. [4]
The worst prognosis cases are those related to cancer (paraneoplastic), male sex, and when there is esophageal and cardiopulmonary involvement. [2, 25]
Liubing Li et, reported that the presence of anti-MDA5 antibody correlates with DM and could be used as biomarkers in the clinical diagnosis of CADM. They also report that their positivity was significantly associated with an increased risk of death, as well as a worse prognosis concerning the overall survival of patients with DM. [28]
Despite all diagnostic tools that we currently have, the diagnosis of dermatomyositis remains a challenge, due to the variability of the disease behavior.
Early intervention can reduce disease-related morbidity, decrease the risk of developing interstitial lung disease, and improve survival in affected people.
In our patient specific case, it may be that some environmental factor has triggered the clinical picture compatible with dermatomyositis associated with anti-MDA-5, it could even be that some metabolite associated with the consumption of hallucinogenic mushrooms has caused abnormal signaling in the via IFN, in particular, Interferon-β (INF-β), where the generation of high levels of IFN can induce auto antigens such as MDA5, activate immature dendritic cells to become antigen-presenting cells, increase expression of major histocompatibility complex class I (MHC I) and activate lymphocytes. [1, 4, 10]
None
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
