
Review | DOI: https://doi.org/10.31579/2640-1053/077
*Corresponding Author: Peng H Tan, Breast Unit, Department of Surgery, Royal Free NHS Foundation Trust, Pond Street, London, NW3 2QG UNITED Kingdom
Citation: M Xie, E Sfakianakis, P H Tan. (2021) Complex interaction of adipokines in breast cancer and anti-tumour immunity; a new paradigm for cancer treatment. Cancer Research and Cellular Therapeutics. 5(2); Doi: 10.31579/2640-1053/077
Copyright: © 2021 Peng H Tan, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium,provided the original author and source are credited.
Received: 08 April 2021 | Accepted: 26 April 2021 | Published: 28 April 2021
Keywords: leptin; adiponectin; breast cancer; therapeutic target
Obesity and its related complications have been the pressing disease pandemic affecting the developed world. It is well-established that the direct consequence of obesity in the cardiovascular system resulting in many diseases. However, its implications in carcinogenesis, cancer treatment and one’s anti-tumour immunity are gradually unfolding. To understand how fat cells can affect these, one needs to explore how the fat cell affects epithelial and immune cells. To this end, we explore the way how the adipocytes, via its production of adipokines, influence these cells, resulting in early epithelial cell transformation into cancer cells and influencing anti-tumour immunity once the cancer is established. In order to simplify our discussion, we focus this review on breast cancer. We propose that to have an effective therapy for cancer treatment, we need to intervene at the adipokine interaction with epithelial cells, cancer cells, and immune cells. In this review we also decipher the potential therapeutic targets in controlling carcinogenesis and disease progression.
Obesity is traditionally viewed as the chronic and excessive growth of adipose tissue. However, it is now gradually regarded as a “global pandemic”, not only affecting the developed world but slowly impacting on the developing world. Its direct consequence on the cardiovascular system has been rather worrisome in many cardiovascular diseases. In this review, we examine its impact on breast cancer (BC) development and the disease progression.
Fat cells, the major constituent of the adipose tissue are no longer viewed as a passive energy store but lately they are regarded as a major complex of endocrine and metabolic organ. To do that, adipocytes express and secrete many key cellular modulators which have a permanent role in the development and regulation for many disease processes. These modulators that are involved in cell-cell signalling proteins are generally termed as adipokines. So far, many adipokines have been discovered; but of the most prominent molecules are leptin [1] and adiponectin (APN) [2] as both seem to adopt an opposite function. The former is regarded as a pro-inflammatory molecule [3] whereas the latter generally assumes the anti-inflammatory spectrum [4]. In this review, we decipher the functions of these contrasting adipokines in BC development and its progression, and also anti-tumour immunity.
Cancer development is closely related to ongoing inflammatory processes on the epithelial cells. The contributory effects of adipocytes via its pro-inflammatory arms have been implicated in many obesity-related cancers such as BC, endometrial, prostate, oesophageal, pancreatic and many haematological cancers. Pro-inflammatory micro-environment is imposed by the immune cells via cytokines production and adipocytes via adipokines, and cytokines and many other soluble molecules such as plasminogen activator inhibitor 1 (PAI1) may have a direct effect in promoting carcinogenesis. In contrary, anti-inflammatory processes may modulate the cancer development and its disease progression. Herewith, we provide evidence demonstrating how leptin and APN can affect BC pathogenesis and its progression. We believe that understanding the process of how these adipokines affect epithelial and immune cells, may allow us to successfully tailor our current therapies in treating BC.
Adiponectin and Leptin Structure and Their Receptors
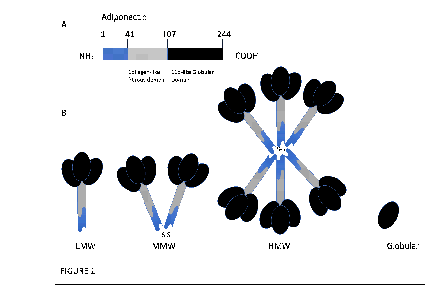
APN is 30-kDa adipocyte complement-related protein (Acrp30) [5] decoded by chromosome 3q27 which is termed as ADIPOQ gene [2]. It has two introns and exons, coding for 244 amino acid long protein, and can be highly polymorphic where there have been 620 reported variants. The structure of the full-length protein (flAcrp) consists of a N-terminal region, a hypervariable sequence, and a collagen-like fibrous domain linked to a C-terminal C1q-like globular domain (gAcrp) (FIGURE 1A). It is also noted to bear some resemblance to complement protein C1q [6]. The low-molecular-weight (LMW) isoform is formed when three monomeric APNs interact between their collagen-like domains (FIGURE 1B). The LMW isoform can further polymerise to form stable multimeric oligomers. Two LMW isoforms can connect via a disulphide bond to form a middle molecular weight (MMW) hexamer, whilst high molecular weight (HMW) isoforms are generated with the help of post-translational modifications to make up larger 12- or 18-mer molecules (FIGURE 1B).
Receptors for APN include AdipoR1, AdipoR2 and T-cadherin. AdipoR1 is found mainly in the skeletal muscles whilst AdipoR2 expression is seen more in the liver [7] although both have been found throughout the body [8]. There is 66.7% homology between these two receptors, which both have 7 transmembrane domain receptors with an internal N-terminal region and an external C-terminal region. They are distantly related to the G protein coupled receptors (GPCR), as it has been shown that their sequence homology to the other GPCR proteins is low [9] and do not seem to be coupled with G proteins. They bind to gAcrp and flAcrp, with AdipoR1 having high affinity whilst AdipoR2 has intermediate affinity.
T-cadherin contains an extracellular part made up of five ectodomains, but unlike other cadherins, it has a GPI anchor anchoring it to the plasma membrane rather than a transmembrane domain [10]. Like AdipoR1 and AdipoR2, it is found ubiquitously in the body but has a particularly high expression in the cardiovascular system on the endothelial and smooth muscle cells [11], with the highest affinity for MMW and HMW isoforms [12]. Its downstream signalling leads to tumorigenesis [13] and during BC there is reduced expression of this receptor [14].
Leptin, belonging to the family of long-chain helical cytokines, was cloned in 1994 which set the milestone in obesity research [1]. It is derived from the Ob/lep gene, located on chromosome 7, which transcribes a 167 amino acid peptide with a molecular weight of 16kD (FIGURE 2A). It is synthesised and secreted mainly by adipose tissues, and therefore, correlated positively with adiposity resulting in most obese subjects have hyperleptinaenia. Circulating levels of leptin communicate the state of energy storage to the brain, hence it is important in the maintenance of energy homeostasis, participating in the anorexigenic pathway through a central feedback mechanism at the level of the hypothalamus.
By alternative splicing of the Ob-R/lepr gene, 6 leptin receptor isoforms (LepRa-f) are generated (FIGURE 2B). These isoforms share a common leptin binding domain but differ in their intracellular domains. Ob (Lep) Ra, b, c, d and f are trans-membrane receptors that all possess the box 1 motif required for binding of Janus kinase-2 (JAK2) [15]. OB-Re uniquely lacks a transmembrane domain hence it exists as a soluble OB-R isoform, binding to circulating leptin in order to inhibit central leptin transport [15].
Of all isoforms, Ob-Rb is central to energy homeostasis and other neuroendocrine functions as it features an extended intracellular signalling domain that is phosphorylated at three distinct tyrosine residues by activated JAK2 (Y985, Y1077 and Y1138) (FIGURE 2). Each of these phosphorylation sites induces a specific signalling pathway with distinct physiological leptin functions. Y985 activates src-homology-2 domain protein (SHP-2) and mitogen-activated-protein-kinase (MAPK) signalling and mediates negative feedback signalling of the leptin signalling pathway. Y1077 activates signal-transducer-and-activator-of-transcription-5 (STAT5) signalling. Finally, Y1138 activates STAT3 signalling (FIGURE 5) and mediates the main effects of leptin on energy homeostasis and neuroendocrine functions [15]. Ob-Rb signalling is negatively regulated by suppressor-of-cytokine-signaling-3 (SOCS-3). SOCS-3 gene expression is increased by leptin-induced pSTAT3 and SOCS-3 peptide binds to Y985 and JAK2 to block leptin signalling in a classic feedback inhibition pathway [16].
Clinical significance of leptin and adiponectin in breast cancer (bc).
Many epidemiology studies have suggested a correlation of BC with leptin and APN (summarised in Table 1). In relation to APN, it has been mostly regarded that there is a higher correlation of BC with lower levels of APN [17], in which it is also associated with greater chance of recurrence [18,19] and a more aggressive phenotype [20-22], hence an inverse relationship of APN level with cancer development [23]. In a similar fashion, APN levels are often higher in non-cancerous tissues [24] and by that reasoning lower in cancerous tissues [25].
It was also found that this inversely related risk is increased particularly in post-menopausal women as compared to pre-menopausal women [26,27] although pre-menopausal women with lymph node metastases also did have lower levels than those without lymph node metastases [28], and higher levels of APN was linked to lower risk of luminal BC in this group. In post-menopausal women, hyperadiponectinaemia reduced the risk of luminal and HER+/neu (human epithelial growth factor receptor) BC [29], and were more strongly inversely related to women who never used post-menopausal hormones and had lower circulating oestrogen levels [30]. Patients with the BRCA1/2 mutations also had lower APN levels, regardless of menopausal status, hence is thought to be associated with early-onset BC [31]. Hypoadiponectinaemia increases the levels of oestrogen and is associated with higher BMI, which might contribute to BC risk [32].
There was also longer survival of patients correlated to hyperadiponectinaemia [33]. Some studies report both LMW and HMW APN correlated positively to a longer survival, and cells also showed reduced viability and proliferation when exposed to APN [34], as anti-proliferative and apoptotic responses in cells are activated [35]. However, another study reports HMW APN showing protective effects for post-menopausal women without a family history of BC or BMI >/=24.0, whilst it was the opposite for women with a family history where APN increases risk of BC [36]. Hence, it may not be as straightforward as it seems since APN comes in different forms that might affect BC risks differently. BC can also induce differentiation of brown adipocytes, rather than white, and since brown ones secrete lower levels of APN [32], it may be why the difference in levels between cancerous and non-cancerous tissues are seen.
Alongside general accepted risk factors for BC, lower APN levels have also been observed in BC patients from Malaysia [37], Taiwan [38], South Korea [39], China [40], and Egypt [41]. Whilst hypoadiponectinaemia increases risk of BC development in most races, there has been studies show that this is more strongly associated in some races than others, for example in Asians more than Caucasians [42].
Epigenetic mechanisms have also been recently shown to affect APN expression. Methylation at CpG sites -74 and -283 nt of ADIPOQ reduces serum APN level, and was increased in BC, giving the low APN levels seen in these patients [41]. Hypermethylation has also been observed in CpG of APN promoter in visceral adipose tissue at positions +128 and +76 from transcription start site [41], as well as in subcutaneous adipose tissue [43].
Hyperliptinaemia has been linked to the formation and development of BC, its aggressiveness and bad prognosis in epidemiological studies [44,45] (TABLE 1). Hyperleptinaemia is positively associated with high BMI [46] and BMI is inversely related to the risk of pre-menopausal BC [47]. Therefore, not surprisingly there is a strong association of leptin with risk of BC [44,48]. This association may be due to the fact BC itself expresses leptin and its receptor [49,50]. Hence, leptin may simply be a marker of disease development [51] so may be used as a screening tool in groups with high BC risk.
In animal models, the incidence of BC is decreased in either leptin deficiency or leptin receptor deficiency [52]. In humans, a meta-analysis showed that 7 out 9 studies without heterogeneity indicates that leptin is strongly correlated to BC risk, regardless of menopausal status [45]. This finding suggests hyperleptinaemia may promote BC. It is also found that lymph node metastasis positive cases displayed higher leptin [53], indicating the intricated role of leptin in progression of BC. Therefore, leptin levels may be used as a predictor for poor prognosis [53,54]. Irrespective of the BMI, either in primary or metastatic BC, leptin and its receptors are correlated well with tumour size and grade [51,55].
Leptin also regulates aspects of reproductive function by directly influencing the release of oestrogen, which stimulates leptin production and leptin promotes oestrogen production peripherally via its ability to stimulate aromatase expression [56,57]. Next, it can then activate oestrogen receptor (ERα), and ERα-dependent transcription in ligand-independent manner [58]. In the presence of the anti-oestrogen treatment (Faslodex), leptin stabilises ERα, interfering with the proteasome-ubiquitin pathways of ERα degradation. Consequently, high leptin can worsen BC progression in patients with ER+ BC [59] and interfere with the successful treatment of anti-oestrogen therapy in BC.
Leptin may also be involved in BC carcinogenesis through cell proliferation or tumour progression as it acts as a growth factor and regulator of cell proliferation [52,60]. At least in a murine model, leptin contributes to and is required for mammary tumorigenesis. Specifically, leptin induced cell growth and stimulated the expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 (VEGF/VEGFR2), of which these can be inhibited by pre-treatment with leptin antagonists [61]. In BC biopsies, leptin and its receptors are overexpressed, whilst it is absent or expressed at very low levels in normal epithelium or benign tumours [50,51]. Leptin activities are mediated through the Ob-Rb that, upon leptin binding, can stimulate the JAK/STAT3, (extracellular signal-regulated kinase) ERK1/2, and phosphatidylinositol-3-Kinase (PI3K) pathways as well as induce cyclin D1 expression and retinoblastoma protein hyperphosphorylation [49,62]. In addition, leptin can also transactivate HER2/neu [63] and induce expression of VEGF/VEGFR2 (FIGURE 4) [64]. All these data suggest that Hyperliptinaemia might impede different BC therapies, including those targeting ERα, human epithelial growth factor receptor 2 (HER2/neu) or VEGFR.
Studies have shown that hypoadiponectinaemia in the background of hyperliptinaemia increases the risk of BC [38,65]. This was also seen in treatment for BC with anti-oestrogen therapies significantly decreasing the leptin/APN ratio, whilst individual level correlations were not always sustained [38,66], and sometimes not seen to be affected at all [67], even though tumour regression was still seen. MicroRNAs (miRs) have also been identified in metabolic diseases and can be deregulated in BC patients. Of note, miR-17-5p was higher whilst miR-221-3p was lower in BC patients compared to controls [68]. miR-17-5p has been shown to promote adipogenesis [69], hence affecting levels of APN and leptin indirectly, thus increasing the risk of BC. miR-221-3p has a more direct effect on APN, where it mimics repressed expression of APN [70].
Biological Effects of Adipokines on Epithelial Cells
To understand biological effects of APN, it is important to define how it interacts with epithelial cells and epithelial-to-mesenchymal transformation (EMT). APN can activate 5'-adenosine monophosphate-activated protein kinase (AMPK), and inhibiting PI3K/AKT (protein kinase B), mammalian target of rapamycin (mTOR), and JAK/STAT pathways. It can also directly inhibit glycogen synthase kinase (GSK)-3β, which is a downstream signalling molecule of Wnt. These then stop cells from surviving or proliferating further, and the key relevant pathways affect in BC biology include Wnt, PI3K/AKT, GSK3β, mTOR, and AMPK [71].
APN is able to modulate the Wnt/GSK3β/β-catenin pathway by reducing serum phosphorylation of GSK3β, resulting in reduced nuclear translocation of β-catenin [72]. The canonical pathway describes in the normal state when a low amount of Wnt is present, GSK3β can induce the ubiquitination of β-catenin [73] (FIGURE 3). In many cancers, Wnt is hyperactivated such that GSK3β is inhibited [74]. This means that there will be more stabilised β-catenin, hence it will be able to translocate into the nucleus and bind to T-cell factor/lymphoid enhancer factor (TCF/LEF), a transcriptional factor, thus activating a cluster of genes that helps the cell establish oncogenicity [75]. When APN is present, GSK3β continues to cause β-catenin ubiquitination, hence transcriptional activity and tumorigenesis decreases [72]. In BC, it was seen that high levels of stabilised β-catenin was seen in at least 50% of patients with clinical disease [76], and it is believed to play a role in promoting triple negative BC (TNBC) and HER2+ BC [77].
The PI3K/AKT/mTOR pathway is another key pathway both directly affected by APN and seen to be altered in BC (FIGURE 3). Upstream to this signalling is the epidermal growth factors receptors (EGFR), including HER2/neu [78], and upon ligand binding it dimerises to lead to PI3K/AKT/mTOR signalling [79]. The focus in cancer is usually alterations to PI3K/AKT signalling and this triggers downstream signalling, including mTOR [80], which enables cell cycle progression, survival and proliferation, leading to tumorigenesis [81]. At signalling interface, this pathway can be inhibited by AMPKα and peroxisome proliferator-activated receptor (PPAR) [82], a phenomenon replicated by APN [7], leading to reduced expression of PI3K/AKT [83]. APN is thought to be important in preventing BC pathogenesis also through the PPAR pathway [84], and APN deficiency increased the PI3K/AKT/β-catenin signalling [85]. Hence, targeting APN that it affects can also play a role in controlling the disease, in particular in HER2+ cancer.
In addition, there is evidence to support that APN can inhibit mTOR phosphorylation directly, arresting cellular growth [86]. mTOR responds to nutritional status, growth factors and stress signals around the cell, then has downstream signalling to regulate between cell growth and cell death. It can also be affected upstream by the PI3K pathway [87]. When activated, mTOR promotes cellular growth by reducing the autophagy machinery [88]. mTOR can activate the hypoxia-inducible factor (HIF-1α transcription factor), such that there is increased glycolysis due to increased expression of relevant enzymes and increased glucose uptake as more GLUT1 transporters are present [89]. This in turn helps with cellular survival. Since APN is able to inhibit mTOR, its anti-tumour properties could be exploited for treatment, where it has already been shown that mTOR inhibition is able to stop cellular growth even if it is further upstream dependent on AKT activation [90]. Several cancers have had trials on mTOR inhibition [91,92]. The pre-clinical trials were promising, but in the clinical stages there was high toxicity burden, and the effect was minimal [93]. Hence, it would be interesting to see if APN affects it in a different way, maintaining the therapeutic effects with minimum toxicity.
APN activates the AMPK pathway [94] and it has recently been observed that AMPK activation plays a role in suppressing inflammasomes that allow BC growth [95]. This pathway is generally tumour suppressive, and helps to modulate inflammation, arrest the cell-cycle and oppose metabolic changes seen in cancers [96]. It also induces apoptosis via p21 and p53 signalling [97], and as discussed earlier has a role in inhibiting the mTOR pathway [98] and hence an effect on autophagy [99]. Another important step in how APN prevents BC is that APN levels are negatively correlated to cell distress and membrane disruption, which when too low can eventually lead to tumour initiation [100]. Direct physical effect on epithelial cell structure on tumorigenesis remains to be in the early stages of our understanding the effect of APN.
One of the peripheral functions of leptin is a regulatory role in the interplay between energy metabolism and the immune system [3], in part responsible for the inflammatory state associated with obesity. Leptin promotes the development and progression of BC by activating the JAK2/STAT3 pathway [101] (FIGURE 5). Upon binding of leptin to its receptor, activated JAK2 phosphorylates the tyrosine 1,138 residue, which acts as an anchoring site for the STAT3 to recruit SH2 domain, resulting in dimerization of STAT3 and its subsequent translocation to the nucleus. STAT3 dimers acts as activators of the transcription of various genes, such as c-myc, cyclin D1, p21/waf1, c-jun, junB, erg-1, and Bcl-2, all of them involved in cell growth and proliferation [102] (FIGURE 5). For example, leptin can regulate the cell cycle and increases BC cell growth by inducing cyclin D1 expression via STAT3 activation [103]. To counter this, this dimer activates the transcription of genes such as SOCS3, which modulates these effects. Enhanced STAT3 signalling leads to altered expression in the key regulators of EMT to augment invasiveness and migration of cells [104]. Another proliferative effect of leptin in BC is via upregulation of human telomerase reverse transcriptase (hTERT) activity [105].
Leptin participates in the expression of stem cell self-renewal transcription factors Nanog such as Sry-related HMG box (SOX2), and octamer-binding transcription Factor (OCT4) [106]. For example, it has been shown that leptin activated STAT3 is pivotal in cancer stem cell (CSC) maintenance in TNBC [107]. Leptin induces canonical Wnt1 signal pathway functioning through β-catenin-dependent mechanisms. Several leptin dependent signalling kinases, including ERK-p90-RSK and Akt, which phosphorylate GSK3β, along with the increase of MTA1 expression, regulate the function of this Wnt pathway to promote EMT in BC [108]. Another complex signalling crosstalk between leptin, Notch and IL-1 seems to be an important driver of leptin-induced oncogenic action, proinflammatory effect and pro-angiogenic state [109].
Regarding angiogenesis regulation, the activation of HIF-1α and nuclear factor kappa B (NF-κB) by leptin via its upstream JAK/STAT3 pathway are essential in the regulation of VEGF (FIGURE 4) [110]. Leptin can participate in the phosphorylation of VEGFR-2 independently of VEGF in endothelial cells and BC cells [111].
Cross interaction of leptin with many pathways can also cause BC carcinogenesis and progression. The interaction of leptin with Transforming Growth Factor (TGFβ1) seem to promote metastasis and CSC related recurrence [112], likely participating in the inhibition of acetyl coenzyme A carboxylase 1 (ACC1) [113]. Bidirectional crosstalk between leptin and Insulin growth factor-1 (IGF-1) signalling resulting in the activation of EGFR promote proliferation and migration of TNBC cells [114]. Not only HER2 can induce the production of leptin by epithelial cells [115], but also leptin transactivates HER2 through the activation of the EGFR and the activation of JAK2, resulting in the growth of HER2+ BC cells [116]. On the other hand, one can argue that leptin may contribute toward to sensitivity of anti-HER2 therapy due to its ability to induce the expression of HER2 by BC.
Leptin can enhance tumorigenicity in ER+ BC, probably by stimulating cellular proliferation via oestrogen and oxidative metabolism mediated by various cytochrome P450 (CYP) enzymes such as CYP1A1 and CYP1B1. A bidirectional interplay between ObR signalling (FIGURE 5) and ERα was suggested by the statistically significant correlation between the expression of both receptors in BC cell lines and ex vivo studies [117]. Leptin directly promotes synthesis of oestrogen by enhancing aromatase expression in BC cells [58] and adipose stromal cells [118] and hence can increase of risk of developing ER+ BC. Alternatively, together with prostaglandin E2 (PGE2), leptin have been shown to drive aromatase expression via the suppression of the metabolic regulators LKB1/AMPK [119]. Leptin-dependent aromatase expression has been correlated with cyclooxygenase (COX-2) upregulation, which is involved in PGE2 synthesis and cooperation among multiple signalling pathways [120]. Furthermore, a novel mechanism has been proposed based on leptin-dependent MAPK signalling, the suppression of p53, and HIF1α and pyruvate kinase M2 (PKM2) as direct mediators of aromatase expression [121].
Oestrogen is a ligand for the ERα and a substrate for CYP1B1. Leptin can induce CYP1B1 expression in ERα+ BC cells in a mechanism that involves AKT and ERK signalling pathways [122]. Leptin also promotes cell viability and proliferation through crosstalk with ERα (FIGURE 5). Both STAT3 activation and ERK1/ERK2 signalling mediated by leptin have been described to act as a key event in ERα-dependent development of BC. Many mechanisms have proposed. One is JAK/STAT3-AKT signalling pathways in the suppression of the extracellular matrix protein CCN5, which acts as an anti-invasive element in BC [123]. Alternatively, ER signalling mediates leptin-induced growth of BC via autophagy induction [124].
Chronic Inflammation and Adipokines in Cancer
Chronic inflammation has been established as a key process in BC pathogenesis and progression [125]. Traditional pro-inflammatory cytokine such as TNF-α and IL-6 have been implicated for poorer BC prognosis [126]. Similarly, leptin imposes pro-inflammatory proprieties like these cytokines [111]. APN has many anti-inflammatory effects.
APN is able to reduce IL-6 and TNF-α secretion by macrophages [127] and endothelial cells [128,129] in the long term. It also reduces the TNF-α secretion by dendritic cells (DC) and T cells [130]. In addition, high APN levels also increases programme death-1 (PD-1) expression on CD8+ T cells, which is associated with immunological tolerance [131]. APN level is inversely correlated to tumour oxidative status [131].
In BC, APN alters signalling pathways by acting on STAT3 and NF-κB. STAT3 is involved in tumour proliferation, survival and invasion, at the same time reducing anti-tumour immunity [132]. APN can specifically affect the STAT3 pathway in macrophages [133] and DC [134], and this is all seen to have a suppressive effect on the NF-κB pathway. STAT3 downstream signalling can then activate NF-κB as well, especially after IL-6 stimulation [135], and they have been seen to both be continuously activated in the same tumour cells [136]. There are a variety of ways that the NF-κB pathway can be suppressed by APN in immune cells in addition to the STAT3 pathway. In macrophages, NF-κB can be suppressed by inhibiting ERK1/2 [137], MAPKp38 [133,137] and c-JNK [133], or by activating AMPK [138]. For DC, NF-κB suppression via STAT3 is activated by AMPK and MAPKp38 pathways from AdipoR1, which also activate the SOCS3 pathway [134], and COX2 and PPARγ activation also suppresses NF-κB [134]. Endothelial cells are seen to have an increasing role in tumour formation, where they suppress the NF-κB pathway by increasing intracellular cAMP and AKT phosphorylation [139], which is able to stabilise IκB such that NF-κB is not activated [140].
Additional evidence that inflammation can cause BC is seen during the current severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic. There have been cases of relapse in remission of BC patients who have had severe coronavirus disease 2019 (COVID-19) that could be attributed to a proinflammatory environment including neutrophil extracellular traps (NETs) [141]. However, this is only a preliminary observation, and more studies would need to be done to solidify the link between COVID-19 and recurrence of BC.
Role of Adipokines in Anti-Tumour Immunity
It is generally accepted now that there is a tumour microenvironment (TME), where the appropriate environment will allow tumour cells to keep proliferating [142]. Of note is the immune microenvironment, where latest findings have shown that it could stop tumour growth and prevent metastases [143]. In many cancers, it has been seen that presence of T cells in the TME has been linked with better prognosis [144]. This is also seen in BC and a highly immune infiltrative environment has been linked to a good prognosis [145]. Increased T cell density in the TME has been associated with an increase in overall survival time for BC patients [146]. Specifically, CD8+ T cells are associated with a good prognosis for ER- and ER+/HER2+ BC [147], which is further evidenced by poorer prognosis for patients with low T cell immune levels [148].
Macrophage infiltration into the TME also affects the progression of the tumour. M1 macrophages are pro-inflammatory, whereas M2 macrophages accelerate tumour growth and tissue remodelling. Benefits of having a TME polarised to M1 macrophages has reduced tumour growth [149], showing that there could be benefits of having a pro-inflammatory environment to increase the anti-tumour effects.
In studies related to BC, it has been seen that genes involved in oxidative stress and T cell aggregation that are seen to dampen growth of the tumour have the highest expression in a high immune infiltration and T cell TME, and this similar environment on the other hand had the lowest expression of genes involved in the Wnt pathway [146] hence not allowing the tumour to grow as the Wnt pathway is inhibited [150]. Several studies have also found that high immune infiltration BC have more TP53 mutations [151], which can disrupt the tumour’s ability to stabilise its genome [152]. However, this effect is reduced in HER2+ BC [153].
APN is usually associated with a reduced cancer risk, however hyperadiponectinaemia could result in tumour progression. The TME seems to favour a pro-inflammatory environment in order to combat the tumour, and although APN is a primarily anti-inflammatory molecule it does have some pro-inflammatory properties. Macrophages are only transiently induced to produce TNF-α and IL-6 [127], activated by STAT-3 [154] or ERK1/2 [155] and Egr-1 [156] activating NF-κB. DC can start producing pro-inflammatory cytokines like IL-6 through activation of NF-κB via Jun N-terminal Kinase (JNK) pathways [157]. T cells are also activated and produce IL-6 [158]. Fibroblasts can also secrete pro-inflammatory cytokines like IL-6 to contribute to the state of inflammation, mediated by activating NF-κB through AMPK, MAPK p38 and ERK1/2 pathways [159,160].
Generally, the lower the level of inflammation, the better it is in terms of preventing tumorigenesis. However, the TME has shown that the right immune cells might need to be present in order for the tumour cells to be cleared. This could also mean that APN is better at preventing BC rather than treating it, and it has been shown where APN signalling to DC could prevent further development of these cells such that there is tolerance to tumour antigen as the antigen presenting cells (APCs) are not priming the T cells for anti-tumour immunity [134]. Tumour cells have also been seen to attenuate natural killer (NK) cell functions and recruit an excess of regulatory T cells, creating a pro-tumorigenic profile that allows the tumour to carry on proliferating unchecked [161]. There is also a very complex network of immune cells and molecules along with signalling pathways that can also be affected by the concentration of APN present, hence more data needs to be obtained before coming to a conclusion about the therapeutic effects of APN on BC. Whereas for leptin, it promotes systemic inflammation via TNF-α and IL-6 [111,162], and induces HIF-1α and VEGF which can propagate cancer survival, proliferation and migration [163][164]. Therefore, modulating chronic inflammation by interfering leptin axis before establishment of cancer is crucial. However, in the TME, the role of leptin promoting anti-tumour immunity can also be denied for successful treatment of BC [165].
Potential Therapeutic Target by Interfering Adipokine Axis
Interestingly, glitazone, a diabetic medication, has been seen to increase APN production and inhibit proliferation of BC cells in vitro [166]. Rosiglitazone can also reverse dexamethasone-induced reduction in APN levels [167]. Another drug liraglutide can also reduce inflammation and is seen to increase mRNA levels of APN, so it is thought that there could be anti-proliferative effects on cancer cells of obese subjects [168]. Symbiotic supplements have also been shown to increase APN levels in randomised controlled trials, as well as reducing proinflammatory TNF-α and CRP levels, hence they might be helpful treatment for patients [169]. Due to the epigenetic mechanisms of suppressing APN in a state of inflammation and in obesity, it has been found that this is via a DNMT1-dependent mechanism as DNMT inhibitors reversed the reduction in APN [170]. There are some APN producing regulatory T-cells in the thymus of mice, and preliminary experiments have shown that expressing this subtype of T cells can inhibit BC development [171].
Different proposed therapeutic strategies include the use of soluble OBR, peptide-based leptin antagonists and ObR blocking antibodies [172]. For example, benzyl isothiocyanate can inhibit oncogenic action of leptin in BC cells by suppressing activation of STAT3 [173]. Apart from this, direct interference to promote leptin negative regulation are emerging as novel therapeutic targets for patients with breast cancer. For example, the anti-leptin activity of vitamin D in oestrogen-sensitive tumours in women seems to mediate hTERT downregulation [174]. Similarly, PPARγ ligands are demonstrated to inhibit leptin signalling mediated by MAPK/STAT3/AKT phosphorylation and counteract leptin stimulatory effect on oestrogen signalling [175]. Thus, PPARγ ligands have been suggested as therapeutic molecules for breast cancer treatment [175].
Physical activity affects both adipokines concentration as well as APN:leptin ratio [176]. It can also moderate inflammation and all these changes can reduce tumour proliferation [177]. Likewise, calorie restriction also influences both adipokines. For example it may also allow higher APN levels therefore reduced tumour incidence and volume [178]
It can be said that different adipokine needs to be targeted differently at different stage of BC. In stage of EMT or perhaps the early BC, promoting the activities of APN and antagonising the action of leptin can generate the favourable outcome in controlling BC. When one is relying on anti-tumour immunity to mount adequate response to cancer, avoiding the use of APN and perhaps introducing leptin either endogenously by increasing BMI or exogenously by pharmacological means may be an attractive approach in ensuring successful treatment of advance cancer.
Obesity is a well-established risk factor for developing BC at least in post-menopausal women. The molecular mechanisms underlying the relationship between obesity and breast carcinogenesis involves adipokines, oestrogens, insulin and inflammatory cytokines. In this review, we showed that activation of leptin signalling results in concurrent activation of multiple oncogenic pathways leading to an increased proliferation, epithelial-mesenchymal transition, migration and invasion of BC cells, whereas APN has the opposite effects. The knowledge of the complex molecular network of leptin and APN signalling responsible for mammary carcinogenesis may provide novel ideas for the prevention and treatment of BC associated to obesity.
Even though the association between obesity, inflammation and BC is clear, some controversial data still remains [164]. The dialogue between the adipocyte and the BC cells is well-known to potentiate not only the growth or invasion, but also treatment resistance [179]. In health, role of mitogenic effect of leptin and anti-mitogenic effect of APN can contribute to breast carcinogenesis. Once cancer is established, most data support that moderately increased BMI may improve survival and response to treatment, but an increase of BMI to morbid level can easily attenuate these benefits; the concept termed as an “obesity paradox” [180].
BMI should be viewed in the context of the stage of the disease, since advanced cancer results in weight loss. Moreover, the increased fat stores may provide an energy reserve that may be useful for a longer survival time. One needs to constantly balance the state of chronic inflammation secondary to obesity via production of leptin and other proinflammatory cytokines which may lead to cancer versus very low BMI with high level of APN which may produce some immunodeficient state that favours the immune escape of cancer cells. It can be argued that the proinflammatory state in advanced cancer may be helpful for the immune response against the tumour. Whether leptin contributes to this favourable effect on the response to immunotherapy in advanced cancer warrants further investigation [181]. Promotion of elevated APN and suppression of leptin in healthy subject may prevent breast carcinogenesis.
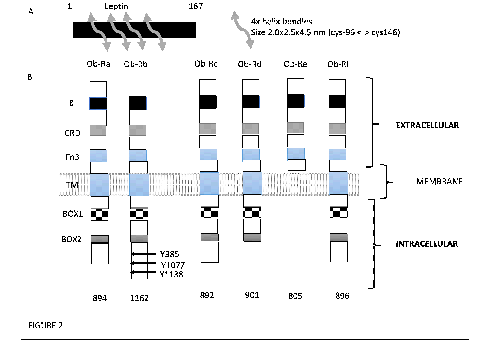
A, Leptin belongs to the family of long-chain helical cytokines, which includes leukaemia inhibitory factor, ciliary neurotrophic factor (CNTF) and human growth hormone, shows the molecular structure of leptin with 4 helical bundles.
B, Leptin receptor (CD295) also known as LEP-R or OB-R is a type I cytokine receptor. Isoforms of Ob-Ra to f are schematically represented. 6 differently spliced isoforms of the ObR have been documented. All the isoforms share identical extracellular binding domains CRD = cytokine receptor domain; Fn3 = type 3 fibronectin domain; Ig = immunoglobulin domain; Box 1-3 – constant intra-cellular motif; Box 1 motif is required for JACK interaction and activation. JAK = tyrosine kinase; STAT = signal transducer and activator of transcription; SOCS = proteins-cytokine signal transduction inhibitors. Three tyrosine residues, whose phosphorylation is important for leptin signalling, are indicated in Ob-Rb: Y985 interacts with the SH2-containing protein tyrosine phosphatase 2, Y1077 with STAT5, and Y1138 (Box3) with STAT3.
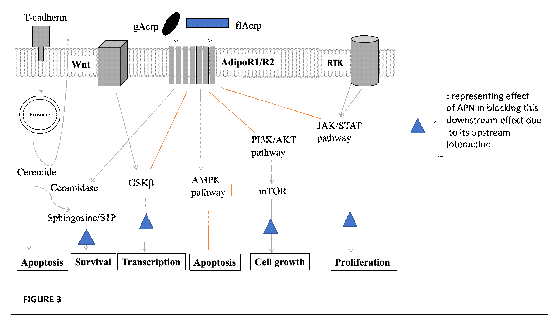
In normal epithelial cells, APN is bound by T-cadherin and presented directly or indirectly to AdipoR1/R2 to inhibit signalling pathways activated in neoplasia. APN activates AMPK, and inhibits PI3K/AKT, mTOR, MAPK and JAK/Stat pathways, or directly affects GSK3β to suppress oncogenic pathways. Cancer cells downregulate T-cadherin while AdipoR1/R2 expression persists, and oncogenic pathways prevail. One model is that ceramidase activity associated with AdipoR1/R2 weighs the balance in favour of cancer cell survival. T-cadherin expressed in the tumour vasculature promotes cancer as a pro-angiogenic factor in cooperation with APN (not shown). Black arrows: activating pathways; Blunt end: inhibitory pathways.
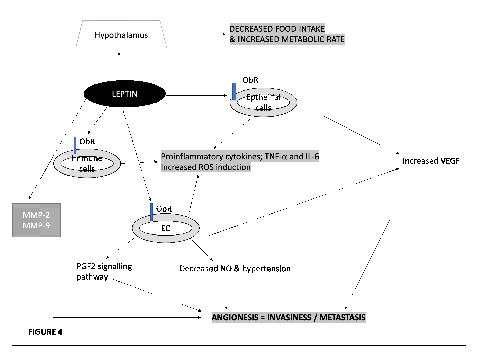
Leptin secreted by fat cells can promotes VEGF resulting in vascular permeability that promotes angiogenesis through vascular fenestration. It also increases proinflammatory cytokines that leads to angiogenesis. VEGF; vascular endothelial growth factor.

Mechanisms of leptin action in breast cancer cells. The cartoon shows the signalling pathways that mediate the leptin effects on breast cancer cells. ObR can couple its outcome with interaction with insulin receptor substrate (IRS) phosphorylation which activates PI3K/AKT pathway through the relationship of IRS with subunit p85 of AKT. AKT phosphorylates downstream XIAP (an individual from anti-apoptosis protein) hence inhibiting its degradation which leads to diminished Capase-3 action and diminished apoptosis.
Table
Epidemiological studies showing the relationship of serum APN or leptin and breast cancer. Many studies show that low levels of APN are associated with increased BC risk and its disease progression. This is highlighted in the “white” coloured rows of the table. However, studies showing no statistically significant correlations are shown in the table rows that are coloured “grey”. In contrary, high level of leptin is associated with increased BC risk and its disease progression. This is highlighted in the “white” coloured rows of the table. However, studies showing no statistically significant correlations are shown in the table rows that are coloured “grey”. CC; case control study.

Due to space restrictions, the authors were able to cite only a fraction of the relevant literature. We apologies to any colleagues whose contribution to this field might not have been appropriately acknowledged in this review.
Conflict of interest: None.
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
