
Research Article | DOI: https://doi.org/10.31579/2578-8868/129
1* I.M.Sechenov First Moscow State Medical University (Sechenov University), Moscow, Russia
2 Scientific Center of Children's Health RAMS of MOH, Moscow, Russia
1 I.M.Sechenov First Moscow State Medical University (Sechenov University), Moscow, Russia
*Corresponding Author: Khachatryan L.G, Department of Children's Diseases of N.F. Filatov Clinical Institute of Child Health of Sechenov First Moscow State Medical University (Sechenov University), +79166943875
Citation: Khachatryan L.G., Lyalina A.A., Kondakova O.B, Tripelets V.M., Tyurina E.N.,Velikoretskaya M.D., Areyan D.E., (2020) Clinical - genetic characteristics of neuronal ceroid lipofuscinosis type 2. J. Neuroscience and Neurological Surgery. 6(4); DOI:10.31579/2578-8868/129
Copyright: ©2020 Khachatryan L.G, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which p
Received: 13 May 2020 | Accepted: 25 June 2020 | Published: 13 July 2020
Keywords: neuronal cerebral lipofuscinosis; children; neurodegenerative disease;epilepsy
The article is devoted to the issues of diagnosis and therapy of one of the most severe degenerative diseases in children - neuronal ceroid lipofuscinosis (NCL). This is a group of inherited neurodegenerative diseases related to lysosomal storage diseases characterized by regression of psychomotor development, resistant epileptic seizures, vision failure up to amaurosis. The morphological basis of NCL types is the accumulation of autofluorescence material in tissues (particularly in the brain), similar in structure to ceroids and lipofuscin, which are related to the “aging” and “wear-and-tear” pigments. To date, we know 14 variants of diseases associated with mutations in 13 genes (PPT1, TPP1, CLN3, CLN5, CLN6, MFSD8, CLN8, KPUR, DNAJC5, CTSF, ATP13A2, CTD7). [4,6,15]. The most common and deeply studied types of NCL are types 1,2,3. According to scientific data, neuronal ceroid lipofuscinosis is the most common neurodegenerative disease associated with epilepsy and an early fatal outcome. The article demonstrates a unique family case with this disease, reports a discussion of issues related to preclinical diagnosis through genetic verification and suggests a necessity for etiopathogenetic therapy. Here we present two children, from one family, a brother and sister. At the time of diagnosis the sister already had a complete clinical picture of the disease and was genetically verified as having NCL type 2. This fact enabled to identify the same disease in her younger brother at preclinical level and to begin his pathogenetic therapy in time. Currently, the treatment of these patients is conducted with the expensive preparation of Cerliponase - alpha (brineura), which is a purified human enzyme obtained through recombinant DNA technology. Brineura is a recombinant human tripeptidyl peptidase-1 (rhTPP1), the main function of which is the cleavage of the N-terminal tripeptides of a wide range of protein substrates. With the example of this family, the dynamics of clinical manifestations in a child with NCL has been demonstrated in detail, and the algorithm of the medical action aimed at leveling off the serious neurological deficit has been shown.
Neuronal ceroid lipofuscinosis (hereinafter referred to as NCL) is a group of inherited neurodegenerative diseases related to lysosomal storage diseases characterized by regression of psychomotor development, resistant epileptic seizures and visual failure, up to amaurosis. The morphological basis of all NCL types is the accumulation of autofluorescence material in tissues (particularly in the brain), similar in structure to ceroids and lipofuscin, which are related to “aging” and “wear-and-tear” pigments [3, 9, 10,14].
The first descriptions of NCL are found in monographs of the nineteenth century. So, in 1826, Christian Stengel described a case of a family disease manifested by epileptic seizures, delayed psycho-speech development, regression of motor skills and progressive visual failure. Later, in 1896, Bernard Sachs proposed the term “amaurotic familial idiocy” for a group of diseases where he included cases of resistant epileptic seizures with an early debut, progressive visual failure, regression of psychomotor development, and an early fatal outcome. In 1903, Batten, based on the clinical picture, and Shoffer (in 1905), based on a pathomorphological study, detected specific changes in the fundus and lipid accumulation in the nervous tissue in some patients. In 1913, Max Bilshovsky described 3 siblings from the same family with seizures, regression of motor skills with a disease onset after the age of 3.5 years, which is now commonly called a late infantile form of neuro-ceroid lipofuscinosis or Jansky-Bielschowsky disease [1,2,3, 5,12,14,15].
In 1963, a group of scientists Shpilmeyer, Harst, Sjowall, Erickson, Zeman and Alpert discovered the accumulation of specific autofluorescence pigments in tissues in some patients with amaurotic familial idiocy and proposed considering it as an independent disease - neuronal ceroid lipofuscinosis. [1,6,11,18,19]. Currently, 14 types of the disease are known.
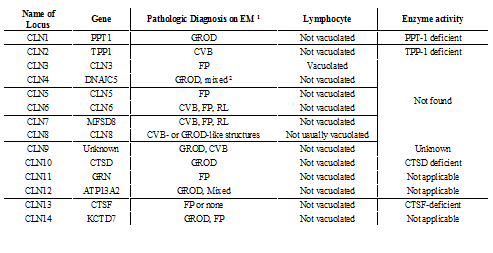
Electron Microscopic Findings and Enzyme Activity by NCL Genotype
(Neuronal Ceroid-Lipofuscinoses Sara E Mole, PhD and Ruth E Williams, MD.[10,13,15,18])
In clinical practice, a different classification is used, and it is based on the age at disease onset which is generally assumed as the delay in psychomotor and speech development, usually preceding seizures [1, 3, 4, 9]:
Congenital form (CNCL)
Infantile form (INCL)
Late infantile form (LINCL)
Juvenile form (JNCL)
Adult form (ANCL)
Northern epilepsy (NE, PEMR)
Among the known mutations in 13 genes associated with the development of various forms of NCL are: PPT1, TPP1, CLN3, CLN5, CLN6, MFSD8, CLN8, KPUR, DNAJC5, CTSF, ATP13A2, CTD7. The most common and deeply studied types of NCL are related to types 1,2,3 [4, 6,10,13,19].
According to world statistics, neuronal ceroid lipofuscinosis is the most common neurodegenerative disease associated with epilepsy and early death [14,19, 20]. To date, hundreds of cases of NCLs of various types are outlined all around the world, especially in the countries of the Scandinavian Peninsula, Italy, Mexico, Japan, but, without doubt, this is only a small part of the undiagnosed cases. The total occurrence of the disease in the world is 1: 25 000. This number varies slightly depending on the region, so in Finland the frequency of NCL is 1: 20,000 covering all the forms. In Pakistan, Romania, the Czech Republic, and Portugal, cases of NCL type 6 are mainly described [2], and for the Russian region the most common type of mutation is TPP1 (NCL type 2). To date, about 120 cases of NCL type 2 have been documented in our country. In all types of NCL, an autofluorescence lipoprotein accumulates in the cell lysosomes. It consists of saposin A and D proteins and / or the mitochondrial ATP synthase subunit [4,5,6]. Pigments with such properties are usually referred to as “aging” and “wear-and-tear” pigments and are called lipofuscin or ceroid [1,3]. Electron microscopy reveals cystosomes filled with aggregates of characteristic morphology and having a specific form of “curvilinear” structures, “fingerprints” or granular osmophilic deposits (in NCL type 1) in the tissues of the nervous system and extraneuronal tissues. The type of these aggregates depends on the type of NCL. Combined types of pathomorphological picture are also possible. Saposins A and D accumulate in congenital and infantile types of NCL. In other forms, the immunochemical basis of the pathology is the accumulation of subunits with mitochondrial ATP synthase [2,4,11,13,19].
According to the literature, the clinical picture of different NCL types is similar at the disease onset. The main clinical symptom is the development of a generalized subtle - clonic (less often an absence seizure) epileptic seizure, which is accompanied by a regression of motor and intellectual skills [5,12,14,21]. Also, a pathognomonic symptom is a progressive vision failure, up to amaurosis, which can be either unilateral (usually at onset) or bilateral. On ophthalmoscopy, retinal pigment degeneration is detected. In NCL type 4, changes in the organ of vision are not observed. The course of the disease is always progressive and has fatal outcome. Each type of NCL has a mapped gene, its own specific manifestation and a course of the disease. NCL type 2 is late infantile type (Jansky–Bielschowsky disease, late infantile form of Batten disease), a disease with an autosomal recessive type of inheritance caused by a decrease in the activity of lysosomal tripeptidyl peptidase. The CLN 2 gene (TPP1) is mapped on the short arm of chromosome 11 (locus 11p15) [4,6,10,15].
Family clinical case.
In April 2017, a 4-year-old girl’s parents brought the child to the children's clinic at Sechenov University with complaints of loss of consciousness accompanied by tonic muscle tension of the extremities with a bite of the tongue and unsteady gait with frequent falls.
According to the anamnesis, the child is from the third, pregnancy without features and the first emergency surgical delivery by Cesarean section, with a long anhydrous interval and intrapartum hypoxia of the fetus. Mass at birth 3980g, length 57 cm. Early motor development proceeded without features. Speech development with a delay, at the moment of examination (age 4 years) with no phrasal speech, expressive speech is in the form of separate words, about ten
The first epileptic seizure occurred at the age of 3,5, in kindergarten, with loss of consciousness, tonic muscle tension and tongue biting. According to the mother, before the attack the girl had received a course of nootropic drugs for the delay of speech development. The child was urgently hospitalized, convulsions were stopped by the diazepam injection. No survey was conducted. A repeated episode of seizures occurred in a month, but without loss of consciousness. Anticonvulsant therapy with a valproic acid was prescribed: Depakine Chrono 300 mg / day. Multispiral computed tomography of the brain revealed a picture of a moderate combined hydrocephalus. Six months later (in the spring of 2017) after an acute complicated respiratory infection and a course of antibiotic therapy, the mother began to notice periodically appearing twitches, tremors in the limbs and discoordination.
According to the survey data:
Blood lactate: on an empty stomach 1.7 mmol / l, after a carbohydrate load - 2.2 mmol / l.
Video-EEG monitoring during wakefullness and sleep revealed moderate brain bioelectric activity disorders. Sleep is cyclical, modulated in phases and stages, physiological patterns of sleep are pronounced. In wakefulness, regional epileptiform activity in the right and left occipital regions, periodically with spread to neighboring areas of the unilateral hemispheres and secondary bilateral synchronization were recorded. In sleep, an increase in the index of the representation of the above epileptiform activity was recorded; single discharges of diffuse epileptiform activity, with an amplitude accent in the frontal parts of the hemispheres were seen.
With the mentioned complaints the child underwent examination at Sechenov First Moscow State Medical University Hospital. At the time of hospitalization the clinical picture of a delay in speech development attracted the attention of clinicians (phrasal speech was absent,there were only 10-15 simple words in the active dictionary, reduced intelligence), marked ataxia, frequent episodes of falls “out of nowhere” were assessed as atonic epileptic seizures. There were no focal symptoms. Muscle tone was hypotonic, tendon reflexes were high, symmetrical. Intentional tremor was not observed.
During the stay in the department, the condition remained stable, and anticonvulsant therapy was selected and administered. While taking topiramate, excitement, tearfulness, frequent falls and tremor of the extremities were noted. So the drug was canceled, and Levetiracetam (Keppra) was prescribed at the dose of 100 mg, followed by an increase of the daily dose to 500 mg (2.5 ml 2 times / day in the morning and evening).
Brain MRI revealed moderate subatrophy of the brain substance, degenerative disease of the nervous system was suspected, and a repeated MRI was recommended to perform in 6-12 months.
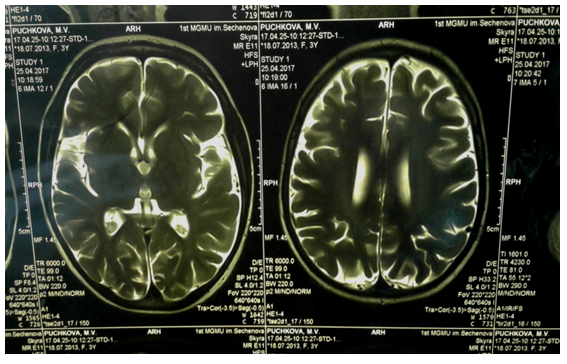
A follow-up examination in a month, while taking anticonvulsants (valproic acid and levetiracetam) according to the recommended scheme, revealed an increase in tremor of the extremities and head, aggravation of ataxia. According to the parents, the girl had more frequent falls. Tremor had a certain kinetics: it intensified in the morning and decreased after lunchtime sleep. The child became lethargic, moody.
Furtherly, the condition of the girl progressively worsened, she stopped walking on her own, difficulties arose with eating and playing (she could not hold a spoon, a toy, etc.). As a result of corrections in therapy, Levetiracetam was gradually canceled. Under suspicion of the metabolic nature of the disease, levocarnitine was prescribed at the dose of 15 drops , 2 times / day.
Three months later, the child was re-admitted to the Department of Psychoneurology of Sechenov First Moscow State Medical University Hospital. The neurological status was as follows: marked emotional lability, delay in speech development, absence of phrasal speech, a decrease in the lexical reserve to 5-7 words, chanted speech. No focal symptoms were detected. In the motor sphere: gait was atactic, walked with support, often fell. Rose independently and stood at the support, but not for long. The girl had tendency of an increased spasticity in the muscle tone of the legs, and hypotension in the arms. Tendon reflexes were high, reflexogenic zones were expanded. Clonuses of the feet on both sides. Marked intentional tremor of the hands during movement, titubation of the head. The central regulation of the pelvic functions was not disturbed, isolated episodes of nocturnal enuresis. The repeated brain MRI and MR spectroscopy (see Figure 2)revealed marked atrophy of substance of hemispheres of the cerebrum and cerebellum, which was assessed as negative dynamics (as compared with the previous research). According to MR spectroscopy, the picture was not specific, the peak of lactate was slightly elevated, which indicated the metabolic nature of the disease.
EEG during wakefulness allowed to reveal a marked neurophysiological immaturity of cortical rhythm accompanied by moderate diffuse cerebral changes in the electrical activity of the brain of residual-organic nature, disorganization and deceleration of BEA. Also, there were signs of a significant increase in the synchronizing effect of nonspecific median structures of the brain . Along with all these, regional epileptiform activity was recorded in the fronto-central region, mostly severe in the temporal area, more often in the right hemisphere. Epileptiform activity in sleep was detected with a low index in the form of regional manifestations in the frontal-central-temporal part, mainly in the right hemisphere.

The conduction of electroneuromyography allowed to reveal changes characteristic of the neuroaxonal demyelinating process. There were no verified changes in the vision.
In view of the disease onset at the age of 3 years with a generalized clonic-tonic convulsive attack and the previous delay in psychomotor and speech development, as well as progressive atrophy of the brain substance according to MRI, signs of axonopathy, increasing ataxia, as well as the transformation of the kinematics of epileptic attacks supplemented by atonic and myoclonic attacks , we suspected neuronal cyroid lipofuscinosis, a congenital degenerative disease of the nervous system which was verified by the results of molecular genetic analysis due to the detection of mutations in the TPP1 \ CLN 2 gene. Subsequently, a survey was conducted with the parents and younger brother of the proband. The results revealed heterozygous carriage in both parents.
The younger brother of proband (aged 1,7 at the time of the molecular genetic study) also had a TPP1 \ CLN 2 gene mutation in a homozygous state without any clinical manifestations. So, an enzymatic diagnostics was carried out in the laboratory of selective screening of the Federal Genetic Center, which showed a sharp decrease in the activity of tripeptil - peptidase in both probands (0.37 μM /hour for a girl and 0.8 for the boy with a normal rate of 7-35 μM / hour ), which also verified the presence of type 2 NCL in both children.
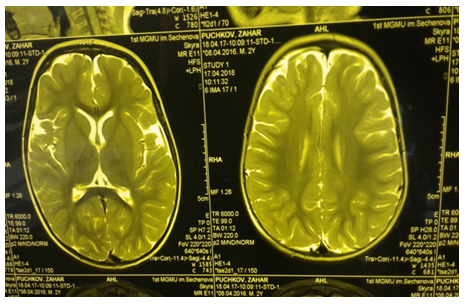
The further dynamic observation showed a progressive loss of motor skills in the clinical picture of the proband (girl), (Figure. 3), a change in muscle tone towards spastic tetraparesis, transformation of epileptic seizures towards the status of myoclonic seizures, permanent large-scale postural and kinetic tremor of hands, head, hyperkinesis of the tongue, stereotypical “Rett-like” “washing movements” in the hands, gradual loss of speech. Because of bulbar disorders and chronic microaspiration, the girl suffered from secondary respiratory complications in the form of bilateral polysegmental pneumonia which was verified at the multispiral CT of the chest. According to brain MRI, negative dynamics was observed in the form of aggravation and progression of atrophy of the brain substance, gross vicarious hydrocephalus (see Figure. 4). Symptomatic, metabolic and anticonvulsant therapy (rivotril and lamotrigine) was administered.


The examination of the proband's younger brother, also having a TPP1 mutation in the homozygous state, revealed no changes on the EEG, ENMG. According to brain MRI data, a minor arachnoid cyst was detected in the left parietal-occipital region, most likely of perinatal origin (Fig. 5). As for the neurological picture of the boy, there was a slight delay in speech development, the motor skills were fully formed. No coordination disorders and convulsive manifestations were noted.
Both children were referred to a concilium of medical experts to resolve the issue of enzyme replacement therapy.
Currently, many scientists in the world are actively searching for specific treatment methods for NCL, since it causes high mortality. One of the achievements of recent years is the research conducted in the USA concerning the use of the Gemfibrozil drug - 5 - (2,5-Dimethylphenoxy) -2,2-dimethylpentanoic acid for the treatment of NCL. The efficacy and safety of the drug was proven by observation of 200 patients with NCL type 2. The therapy was well tolerated by the patients; side effects were observed in only 3 patients, in the form of muscle pain in two patients, and bullous dermatitis in 1 patient. Gemfibrozil is an oral drug that has hypocholesterolemic and lipid-lowering effects and initially has indications for use in hyperlipidemia type IIb, IV, V [8].
According to the results of this study, gemfibrozil can be used in complex long-term treatment as an oral drug in NCL type 2 in children.
In 2017, clinical research was completed ahead of schedule in the United States and the FDA approved cerliponase alfa manufactured by BioMarin Pharmaceutical company as an enzyme replacement therapy drug for the treatment of NCL type 2. Cerliponase-alpha is a purified human enzyme obtained through recombinant DNA technology in the Chinese hamster ovary cell line. Cerliponase-alpha (brineura) is a recombinant human tripeptidyl peptidase-1 (rhTPP1), lysosomal exopeptidase [11, 16, 17, 20]. The main function of the enzyme is the cleavage of the N-terminal tripeptides of a wide range of protein substrates (official guide to brineura).
The prognosis of the disease is unfavorable. Mortality from different forms of NCL varies from several hours (with a congenital form) to several years from the moment of the disease onset. For NCL type 2, the average age of death is 10-15 years.
In families with detected NCL, prenatal diagnosis is available through biochemical diagnostic methods, as well as DNA diagnostics at 11 weeks of gestation, provided that the proband genotype is known. Thus, in families with NCL, it is always necessary to conduct a medical genetic examination and which will help prenatal diagnosis of subsequent pregnancies, as well as to conduct a conversation with parents about the chances of having healthy children.
NCL type 2 is an inherited neurodegenerative disease related to lysosomal storage diseases, characterized by regression of psychomotor development, resistant epileptic seizures, vision failure, even amaurosis. The morphological basis of all NCL types is the accumulation of autofluorescence material in tissues (particularly in the brain), similar in structure to ceroids and lipofuscin, which are related to the “aging” and “wear-and-tear” pigments. The debut of the disease occurs at the age of 2 to 4 years, most often with a generalized tonic-clonic epileptic seizure, which is preceded by a delay in psychomotor and speech development. Children have progressive intellectual impairment, ataxia, regression of previously acquired motor skills with the formation of spastic tetraparesis, increased and modified convulsive attacks (supplemented by myoclonic, atypical absences, focal attacks, etc.), as well as loss of vision up to blindness by the age of 4-5. Knowledge and awareness of the clinical manifestations of this disease, the ability of clinicians to correctly identify it will allow to timely perform administration of pathogenetic therapy and improve the prognosis of these patients, as well as prevent the birth of other sick children in these families, due to targeted prenatal diagnosis.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
