
case report | DOI: https://doi.org/10.31579/2690-4861/684
1Neonatology Department at Hospital General de Castelló, Spain.
2Neonatologist ICGON. Hospital Clinic. Barcelona Spain.
*Corresponding Author: Gemma Arca, Neonatologist, ICGON, Hospital Clínic, Barcelona, Spain. NeNe Foundation, Madrid, Spain.
Citation: Inma Cubells, Gemma Arca, (2025), Characteristic Electroclinical Phenotype in a Newborn with Channelopathy, International Journal of Clinical Case Reports and Reviews, 23(4); DOI:10.31579/2690-4861/684
Copyright: © 2025, Gemma Arca. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 14 January 2025 | Accepted: 04 February 2025 | Published: 20 February 2025
Keywords: neonatal seizures; channeolopathy; KCNQ2 gene; aEEG
Full-term newborn who at 18 hours of life had two episodes of generalized hypertonia and perioral cyanosis lasting 30 seconds, followed by hypotonia and weakness. On physical examination between episodes, patient was drowsy and presented jerky movements induced by acoustic and tactile stimuli. Infection screening test was negative. Ammonia and lactic acid values were normal. No cerebrospinal fluid nor cranial abnormalities were identified. Cerebral function was monitored by amplitude integrated electroencephalogram in which a mirror pattern with descent in the upper and lower margins and presence of monomorphic waves in the EEG were observed, coinciding with episodes of desaturation, cyanosis, and rhythmic diaphragmatic movements. Treatment with phenobarbital was initiated, and later levetiracetam was later added with no response. A metabolopathy was suspected, for which oxcarbazepine was administered, with resolution of clinical symptoms. Clinical exome sequencing confirmed mutations of the SCN1A and SCN2A genes.
An 18-hour-old female newborn was brought to our centre for examination. Patient had had two episodes of generalized cyanosis and hypertonia lasting 30 seconds followed by hypotonia and weakness. Infant was born to a 42-year-old primigravida mother with no relevant past medical history. Pregnancy had been monitored, serological tests were negative (HIV, HCV, HBV), and ultrasounds normal. Streptococcus group B had been found during pregnancy from a vaginal-rectal smear test. An emergency caesarean section was carried out at 38 weeks of gestation due to abnormal echocardiogram. Reanimation was not required. Apgar score of 8/9/10, pHaU 7.36.
On arrival of the newborn to the hospital, neurological examination revealed neurobehavioral alterations: visual inattention, weak cry, and drowsiness, with adequate nociceptive response. Scarce general movements were seen, with scant complexity and variability. Presence of jerky movements in response to acoustic and tactile stimuli, some hyperexcitability, and hyperreflexia of the patellar tendon reflex. Cutaneous pallor and slow capillary refill stood out. The rest of the exploration was anodyne.
Screening blood test to check for infection proved normal; ammonia levels were 100 micrograms/dL; Neither hepatic nor renal alterations, hyperlactacidaemia, or metabolic acidosis were present. Lumbar puncture showed cytochemistry and neuron-specific enolase (21 ng/ml) within normal parameters. Cerebrospinal fluid amino acids, urine organic acids, and plasma amino acids were normal. No abnormalities were found on cranial magnetic resonance and ultrasound scans.
Brain function was monitored with amplitude integrated electroencephalography (aEEG). A pattern of depression in upper and lower margins were observed, coinciding with episodes of desaturation, rhythmic diaphragmatic movements, and stare. Monomorphic waves were observed in the EEG, suggesting electric crises. Treatment with phenobarbital was initiated. Symptoms persisted and levetiracetam was added with no clear improvement. Cyanosis made us suspect of channelopathy, and the patient was initiated on oxcarbazepine, with a favourable initial response.
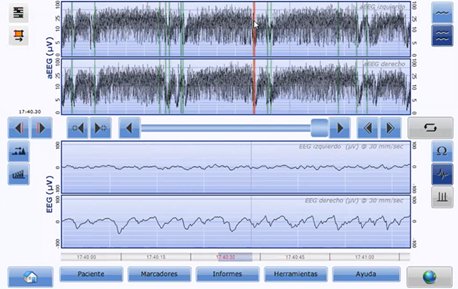
Figure 1: Two-channel aEEG at five days of live. Descent in upper and lower margins are observed with monomorphic waves in the EEG (red line).
Differential diagnosis:
Patients with neonatal seizures and progressive clinical deterioration with suboptimal inter-crises level of consciousness, i.e., neonatal seizure with encephalopathy. Hypoxic ischemic encephalopathy was ruled out in the differential diagnosis because the neurological examination carried out during the first hours of life was normal, Apgar score correct, and pHaU normal. Blood and CSF cultures were negative, excluding infection. No abnormalities were seen in cranial magnetic resonance imaging results, thus, cortical developmental malformations were discarded. No hyperlactacidaemia, metabolic acidosis, or hyperammonemia. Organic acids and amino acids were negative. A microarray was requested, with a normal result. Clinical exome sequencing revealed de novo heterozygous mutations in the SCN1A and SCN2A genes.
Outcome:
At discharge from the neonatal unit, a multidisciplinary team, including neonatologists and neurologists, carries out a follow-up. Five days after discharge from the hospital, seizures resumed -around five crises per day, manifested with generalized hypertonia, medial disconnection, and facial cyanosis. Higher dose of oxcarbazepine was prescribed. However, valproic acid and pyridoxine were later required, with good response. During the first six months of life, she was admitted five additional times, one to the intensive care unit due to status epilepticus. Improvement in the number of crises, with remission at around age of nine months. Patient presents global developmental delay, with difficulties making physical contact with her parents, language, and communication.
Vilan et at. describe a distinct aEEG pattern in newborns with epilepsy associated to mutations of the KCNQ2 gene, which codify for voltage-gated potassium channels. The pattern described for our patient is similar to that reported by these authors.
After the diagnosis (clinical and through aEEG) of a neonatal seizure, identifying the underlying cause is key to provide accurate treatment, minimize epileptic load, and improve long-term neural development. Neonatal seizures are a neurological emergency that require urgent etiological treatment to limit cerebral damage the epileptic load may cause to the developing brain. Presence of cyanosis, apnoea and stare, as in the case of our patient, must alert physicians on the presence of possible tonic seizure, which may be secondary to a channelopathy. Electric crises are usually seen in aEEGs as elevations in the upper margin accompanied, in many occasions, by an elevation in the lower margin. In our patient and in cases of epilepsy associated with mutations in the KCNQ2 gene, descent in the upper and lower margins are observed. In the EEG, monomorphic waves are observed suggesting electric seizure. Rapid detection of the electroclinical pattern compatible with a potential channelopathy may help establish treatment with channel blockers at early stages. The gold standard in the diagnosis of convulsive crises is conventional electroencephalogram. However, aEEG plays a very important role in the diagnosis and treatment of neonatal seizures.
The presence of descent in the upper and lower margins in the aEEG is suggestive of channeolopathy. It may be associated with mutations in KCNQ2, SCN1A or SCN2A genes.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
