
Case Report | DOI: https://doi.org/10.31579/2690-4861/465
1Consultant Division of pediatric orthopedics, Tainan Municipal An-Nan Hospital, Chinese Medical University, Tainan, Taiwan.
2Professor Craniofacial Center, Taipei Medical University Hospital, Taipei, Taiwan.
3Professor Department of pediatrics and medical genetics, National Taiwan University Hospital, Taipei, Taiwan.
4Chief Department of Medical Imaging, Chi-Mei Medical Center, Tainan, Taiwan.
*Corresponding Author: Lin-Show Chin, Consultant Division of pediatric orthopedics, Tainan Municipal An-Nan Hospital, Chinese Medical University, Tainan, Taiwan.
Citation: Lin-Show Chin, Philip Kip-Ting Chen, Ni-Chung Lee, Tsyh-Jyi Hsieh, (2024), Challenge of Clinical Diagnosis and Treatment for Proteus Syndrome: A Case Report, International Journal of Clinical Case Reports and Reviews, 17(3); DOI:10.31579/2690-4861/465
Copyright: © 2024, Lin-Show Chin. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 06 May 2024 | Accepted: 17 May 2024 | Published: 31 May 2024
Keywords: Proteu syndrome; craniofacial dysmorphism; asymmetric overgrowth; hypermenorrhea; whole exome sequencing; whole genome sequencing
Proteus syndrome (PS) is an extremely rare complex disorder, and caused by PS associated somatic mosaic AKT1 gene mutation. PS is characterized by progressive asymmetric, disproportionate overgrowth of patient’s skin, soft tissue, and bone of the different body parts since infant period. There is diversity in both clinical appearances, and with or without PS associated pathogenic genetic mutation situation can be existed for each patient. That caused clinical challenges for both accurate diagnosis and proper early treatment. We report a case of 17 years-old girl she presented with significant craniofacial dysmorphism, including prominent craniosynostosis and generalized skull bone sclerotic hyperostosis changes, hearing impairment after age of 3 years-old, progressive asymmetric overgrowth of the left lower limb with leg length discrepancy in childhood. Focal lipomatous tumors grow over the abdomen, left thigh and spinal deformity developed around teenage years. Hypermenorrhea with adenomyosis and ovarian cystadenoma lesions after menarche 12 years-old. Patient was fulfilled positive clinical criteria > 17 points for clinical Proteus syndrome diagnosis, clinically strict met (mosaic distribution of lesions, sporadic occurrence, and progressive course) three general criteria for PS diagnosis. Although the whole exome sequencing test from affected thigh lipoma did not identify pathogenic/likely pathogenic variant AKT1, PTEN, PIK3CA gene. Whole genome sequencing test from affected cranial hyperostosis bone revealed MSH6 variant of uncertain significance (NM-000179.3: c.754T>G) gene mutation. The patient has received surgical treated at three different medical centers in Taiwan for craniofacial dysmorphism, hearing loss, leg length discrepancy, and hypermenorrhea problems respectively. All clinical symptoms and problems with postoperative have partial relieved and improvement. Further more complicated surgery was necessary for her. Clinically we are challenged both for early confirming PS clinical diagnosis, and make the differential diagnosis from other clinical mimic overgrowth spectrum disorder.
proteus syndrome (PS).
Proteus syndrome (PS), an extremely rare syndrome in the majority of patients, caused by somatic mosaic pathogenic variant AKT1 mutation. PS was first described by Cohen and Hayden in 1979, with an estimated prevalence around 1/1,000,000 liver births. PS patient characterized with post-natal asymmetric, progressive, disproportionate segmental overgrowth of the limbs. Common affecting the skin, soft adipose tissue, and skeleton of the body. Most PS patients present with normal facial appearance and without systemic anomalies at birth, developed progressive asymmetric overgrowth after infant period, which can cause significant craniofacial dysmorphism, severe focal overgrowth, leg length discrepancy, spinal deformity complications. There is clinical diversity between PS patients present with different phenotype severity, and genotype difference- with or without PS associated gene mutation. The PS diagnosis is established in a proband with all three general criteria (mosaic distribution of lesions, sporadic occurrence, progressive course). Patients under clinical score < 10>15 points individual without mosaic AKT1 pathogenic variant condition. Molecular genetic tests play a crucial role in both confirming diagnosis, and make differential diagnosis with other syndromes or skeletal dysplasia. Surgical treatment of symptomatic PS patients including: plastic surgery for craniofacial dysmorphism and lipomatous overgrowth. Orthopedic surgery for leg length discrepancy, macrodactyly, and scoliosis. Dermatologic management for foot cerebriform nevus. Cardiovascular surgery for deep vein thrombosis and pulmonary embolism [7].
These 17 years-old girls was referred from outside hospital to Tainan Municipal An-Nan Hospital Orthopedics Department, for the purpose of tertiary consultation for progressive craniofacial dysmorphism associated with vision and hearing impairment problems. According patient’s parents’ statement she was a G2P1baby, birth date: 2007/05/24, gestation age: 40 weeks, NSD, birth height: 51 cm, birth weight: 3185 gm, head girth: 34 cm, Apgar score: 9 at one minute, 10 at five minutes, birth without obvious systemic deformity condition. Sonogram of the brain was normal at birth, with psychomotor developmental delay. Family history was non-contribution. Mild craniofacial deformity was noted by parents since the age of 2 years-old. 3D-CT image of skull revealed metopic ridge, early closure of anterior fontanelle and trigonocephaly pathologies (Figures 1A and 1B) findings at that time. Under the diagnosis of metopic craniosynostosis she received cranioplasty treatment at CGMHT P-S department aged 2+7 yrs, 2009/12/28. After the age of 3 yrs she developed slow progressive, asymmetric, segmental left lower limb overgrowth clinical symptoms, but with prominent craniofacial dysmorphism (Figures 2A-D) and associated bilateral ear hearing impairment symptoms. First menstruation at age of 12 yrs, associated with hypermenorrhea and absence of pubic and axillary hair development (Figures 3A and 3B), and spinal scoliosis problems (Figures 4A and 4B). The major operative date with Proteus syndrome related surgeries were summarized as follows. [1] PDA s/p transcatheter coli embolism treatment at CGMHT pediatric cardiology 1+3 yrs, 2008/09/24. [2] Metopic craniosynostosis s/p cranioplasty at CGMHT plastic surgery department 2+7 yrs, 2009/12/28. [3] Left leg length discrepancy 4.5 cm (2) s/p guided growth treatment (Lt distal femur and proximal tibia Orthofix 8-plate epiphysiodesis) (Figures 5A and 5B) at NTUHCH orthopedics department 14 yrs, 2021/07/05. [4] Bilateral ear canal stenosis (Figures 6A and 6B) with left ear cholesteatoma s/p left ear canalplasty + tympanoplasty + mastoidectomy+ cholesteatome excision treatment at NTUH ENT department 14+6 yrs, 2021/11/25. [5] Malocclusion, dental caries, gingival hyperplasia s/p tooth extraction, complicated odontectomy, gingivectomy at TMUH oral surgery department 15+3 yrs, 2022/03/22. Pathology report: gingival-squamous hyperplasia, alveolar bone- fibrotic osseous tissue. [6] Adenomyosis uterus with hypermenorrhea s/p hysteroscopic endometrial ablation + adenomyosis excision (Figures 7A-C) treatment at NTUH GYN department 15+8 yrs, 2022/08/22. Pathology report: endometrial hyperplasia with inactive endometrial glands and thickened submucosal tissue. [7] Left inner thigh lipoma s/p excisional biopsy at NTUHT 16 yrs, 2022/12/09. Pathology: Lipoma [8] with whole exome sequencing test. (8) Progressive and recurrent of skull hyperostosis lesions s/p forehead and orbital floor lesions partial resection at TMUH plastic surgery department 16+7 yrs, 2023/11/20. Pathology report: mature trabecular bone, with whole genome sequencing test. Postoperative patient’s problems such as: craniosynostosis, left ear hearing impairment, oral malocclusion, left leg length discrepancy, hypermenorrhea, all with partial clinical improvement. The most recent outpatient patient’s four complaints were: 1. Slow progressive craniofacial dysmorphism with neurological deficits including bilateral hearing, vision and speaking impairments. 2. 2nd sexual development delay with hypermenorrhea. 3. Overgrowth lipomatous tumors over abdomen and left lower limb. 4. Residual leg length discrepancy 2 cm. Physical examination showed significant craniofacial dysmorphism with diffused overgrowth protruding bony nodular lesions over frontal, parietal, and temporal region of the skull, hypertelorism, depressed nasal bridge, and open month disfigurations. Bilateral ear hearing loss, back hyperpigmentation skin lesion with scoliosis spinal deformity, lipomatous overgrowth tumors lesion over abdomen and left lower limb [4], residual leg length discrepancy 2 cm Lt > Rt, absent of axillary and pubic hair. Radiological Imaging both skull films and 3D-CT showed diffuse sclerotic hyperostosis lesions over the cranial outer table regions, and narrowing of bilateral auditory canal (Figure. 8A, 8B, 8C, 8D). Scanogram of spine showed thoracolumbar junctional scoliosis with lumbar megaspondyly changes (Figure.4A and 4B). Triple film of lower limb showed long bones diaphyseal dysplasia change with residual leg length discrepancy post left knee tethering growth surgery (Figure.5A and 5B). Recurrence of hearing loss of left ear, and Impairment of bilateral vision acuity was strongly complained by the patient herself recently.
Written informed consent was obtained from the patient’s parents regarding the publication of the patient's details and associated images.
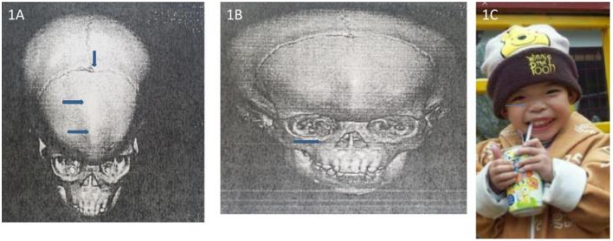
Figure 1A:3D-CT metopic ridge, small anterior fontanelle, 2 years-old. 1B. 3D-CT trigonocephly, midface hypoplasia, 2 years-old. 1C. Gross photo 2 years-old.
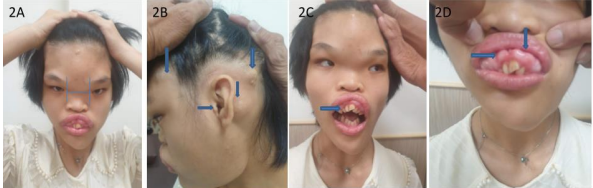
Figure 2A: Gross craniofacial dysmorphism with hypertelorism, sunken eyes, midface hypoplasia, 16 years-old. 2B. Hyperostosis bulging out skull lesions, external ear canal narrow. 2C. Open mouth deformity, crowding tooth, malocclusion. 2D. Gingival hypertrophy, crowding tooth.
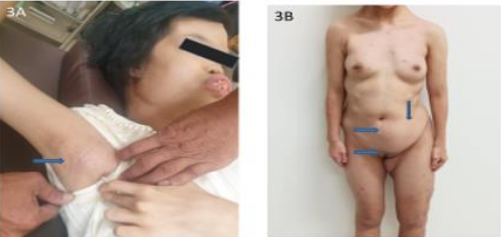
Figure 3A: Absent of axillary hair, 16 years-old. 3B. Absent of pubic hair, abdominal soft tissue mass lesion, 16 years-old.
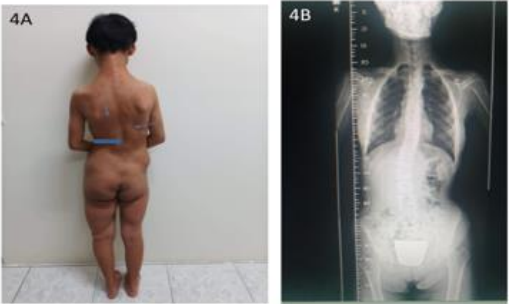
Figure 4A: Scoliosis spine deformity, back skin hyperpigmentation lesions, 15 years-old. 4B. Triple film spine showed Rt thoracolumbar junctional scoliosis, megaspondyly changes of lumbar spine.
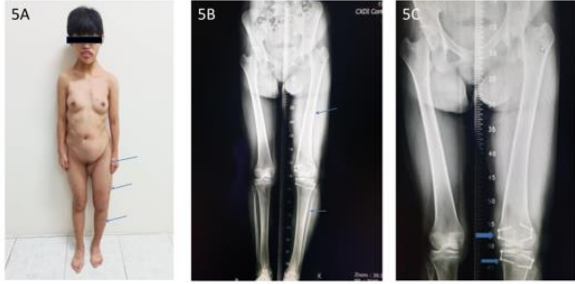
Figure 5A: Disproportional overgrowth with leg length discrepancy Lt lower limb, 16 years-old. 5B. Triple film lower limb showed hypertrophy Lt femur and tibia. 5C. Post guided growth treatment of Lt distal femur and proximal tibia with 8-plate
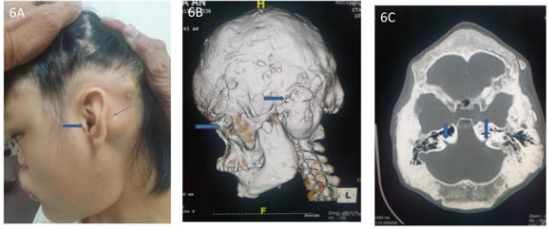
Figure 6A: Gross narrowing of external auditory canal, 16 years-old. 6B. 3D-CT hyperostosis lesion with neural eye and ear compression. 6C. CT Narrowing of bilateral internal auditory canal.
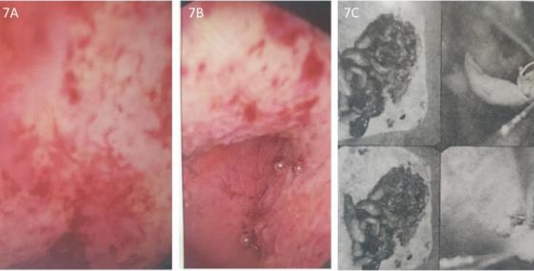
Figure 7A. and 7B: Hysteroscopy generalized endometrial hyperplasia and thickening changes. 7C. Focal submucosal adenomyosis lesions.
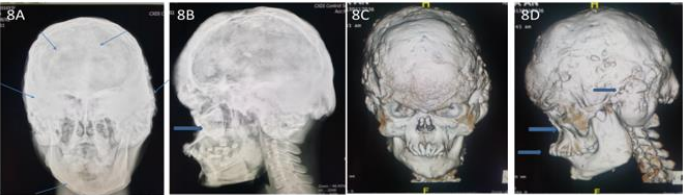
Figure 8A: Skull A-P diffuse sclerotic hyperostosis lesions over cranial outer table region, 16 years-old. 8B. Skull-Lat midface hypoplasia. 8C. 3D-CT severe craniofacial dysmorphism with hyperostosis of skull. 8D. 3D-CT external auditory canal narrowing, midface hypoplasia, and malocclusion
Proteus syndrome (PS) is an extremely rare syndrome characterized by progressive, asymmetric, segmental or patchy overgrowth, commonly affecting the skin, soft tissue and bony tissue. Clinically more than 80% PS patients present with positive sporadic somatic mosaic pathogenic variant AKT1 gene mutation. Most PR patients developed progressive focal tissue overgrowth and caused body disfiguration since the infant period. There are clinical characteristics and three general criteria for PS - With mosaic distribution of lesions, sporadic occurrence, progressive in clinical course. Because of rare prevalence around 1/1.000.000 live birth, medical staff usually lack clinical experience about PS. Combined with the disparity in clinical appearance variation, and with the possibility of either positive or negative pathogenic variant AKT1, PTEN genes genetic identification. Early established “Proteus syndrome” diagnosis is very difficult and challenging for us. For the girl our clinical diagnosis was revised to “Proteus syndrome” after overseas tertiary consultation. Differential diagnosis including- Craniodiaphyseal dysplasia, McCune-Albright syndrome, Neurofibromatosis type 1, PIK3 CA-related overgrowth syndrome. Further Genetic tests- First one with Whole exome sequencing, from left thigh lipoma did not identify pathogenic/likely pathogenic variant on AKT1, PTEN, SOST, GNAS, NF1, PIK3CA gene [6,10]. Secondary one with Whole genome sequencing, from cranial sclerotic hyperostosis bone revealed a MSH6 variant of uncertain significance (NM-000179.3: c.754>G; NP-000170.1: p.Ser 252Ala, heterozygous), and other three carries on GJB2, AKA9, NDUFS2, and RPGRIP gene that cannot explain patient phenotype. Finally, we make the “Proteus syndrome” clinical diagnosis based on her presence with 3 general criteria- mosaic distribution of lesions, sporadic occurrence and progressive course. The patient also had identified 17 points of specific criteria for “proteus syndrome” diagnosis [9,10] including- hyperostosis of skull 5, gingival overgrowth 5, dysregulated adipose tissue 2, linear epidermal nevus 2, specific tumors ovarian cystadenoma 1, facial dysmorphism 2. Although secondary WES and WGS test for PS- associated pathogenic variant did not identified, but we have confidently rule out Craniofacial dysplasia, McCune-Albright syndrome, Neurofibromatosis type 1, PIK3CA-related overgrowth syndrome diseases possibility after detail molecular genetic test. So, we can reasonably establish a “Proteus Syndrome” diagnosis for this patient [3,4,11], and set up our future treatment plans.
Proteus syndrome (PS) is an extremely rare genetic background disorder, manifested with postnatal progressive, asymmetric, disproportionate overgrowth of the body tissue in a mosaic distribution pattern. Clinically majority PS patients were present with syndrome-associated AKT1, PTEN pathogenic somatic gene mutation. PS patients without early diagnosis and treatment, severe complications such as severe craniofacial dysmorphism with cranial nerves entrapment, leg length discrepancy and scoliosis of spine, deep vein thrombosis and pulmonary embolism can happen [7]. Detailed medical history taking, complete physical examination, specific imaging study, affected tissue pathology assessment all are essential for the” Proteus syndrome” diagnosis. Molecular genetic test plays an important role for early confirming PS diagnosis, and help for differential diagnosis. After going through a complete retrospective, I reviewed all sequential hospitalization medical records, and two recent molecular genetic test results (Affected left thigh lipoma whole exome sequencing test and affected skull hyperostosis bone whole genome sequencing test). We revise the clinical diagnosis from MuCune-Albright syndrome and Craniodiaphyseal Dysplasia to Proteus syndrome. Concerned about the future clinical progression, and possibility of foal recurrence problems. We will arrange regular multidisciplinary approach treatment for her. However, clinical challenges still persist with: PS patient’s Genotype Variation - New or Unknown PS gene existing? How to detect and identify? How to completely rule out Phenotype Overlapping and mimic diseases? All puzzles need advancing research and further investigation.
Assistance in clinical Consultation: Jae-Joon Cho, Professor & Chief Division of Paediatric Orthopedics, Seoul National University Hospital, Seoul, Korea.
Assistance in Genetic Assessment: Ni-Chung Lee, professor, Department of Pediatrics and Medical Genetics, National Taiwan University, Taipei, Taiwan.
Assistance in Radiological Interpretation: Tsyh-Jyi Hsieh, MD, Chief Department of Medical Imaging, Chi-Mei Medical Center, Tainan, Tainan
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
