
Case Report | DOI: https://doi.org/10.31579/2637-8892/014
*Corresponding Author: Hathor Hussein, Maadi Psychology Center,Egypt.
Citation: Hathor Hussein, Mehrit Ibrahim (2017) Cerebral Amyloid Angiopathy: An elderly patient with a past history of lobar brain hemorrhage and recent history of recurrent lobar brain hemorrhages. J Psychology and Mental Health. 1(3); DOI: 10.31579/2637-8892/014
Copyright: © Hussein. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 05 July 2017 | Accepted: 18 July 2017 | Published: 28 July 2017
Keywords: intracerebral hemorrhage; amyloid angiopathy; vasculitis; amyloid associated angiitis; caa-related inflammation; steroid and immunosuppressive therapy
Background: Cerebral amyloid angiopathy (CAA) is an important cause of intracerebral hemorrhage. Brain amyloid-related imaging abnormalities (ARIA) can be classified into two groups; those with edema or sulcal effacement (ARIA-E) and those with cerebral microbleeds or small hemorrhages (ARIA-H). There is an aggressive form of CAA, called CAA-related inflammation (CAA-ri) which is characterized by vascular or perivascular inflammation. Current treatment guidelines do not recommend routine use of steroids for intraparenchymal hemorrhage.
Methods: We describe the clinical course, radiologic and pathologic findings of a 78-year-old man with recurrent intracerebral hemorrhage. Diagnostic studies included CT and MRI-scans of the brain, histopathology studies and autopsy.
Results: The patient was diagnosed with a vasculitic form of amyloid angiopathy. Treatment with steroids resulted in clinical and radiological improvement.
Conclusions: There might be a benefit of steroid or other immunosuppressive therapy in some patients with recurrent lobar hemorrhage related to CAA. Patients demonstrating edema out of proportion to the size of hematoma and extension of edema to the sub-cortical U fibers on imaging studies due to underlying vasculitis or CAA-ri may be candidates for such therapeutic interventions.
Cerebral amyloid angiopathy (CAA) is an important cause of intracerebral hemorrhage resulting, from the deposition of amyloid protein in the walls of small and medium sized cortical and leptomeningeal arteries [1]. It was first observed by Oppenheim in 1909 [2] and formally described by Scholz in 1938 [3]. The incidence of CAA is 20/100,000 in persons more than 60 years of age, and there is no difference in occurrence between genders. CAA accounts for up to 10-15% of cerebral hemorrhages in the elderly [4]. There are two different types of CAA; sporadic- and familial forms. The sporadic type of CAA is more common than the familial type and mostly results from the deposition of Aβ peptide in the vessel wall, resulting from cleavage of amyloid precursor protein (APP) by β and γ-secretases [5]. Deposition of these amyloid proteins into the cerebrovasculature is associated with heat shock proteins that trigger release of IL-8, leading to a cerebral inflammatory reaction [6]. Amyloid proteins may also trigger activation of the complement system, alteration of blood-brain barrier permeability, oxidative stress, formation of ion-like channels, and cell toxicity [7,8]. Though the pathogenesis is not fully understood, it is thought that these changes weaken the vessel wall, increasing susceptibility to rupture and hemorrhage [6,9]. The pattern of cerebral hemorrhage on brain imaging studies and histopathological findings are of importance in the diagnosis of CAA.
A 78-year-old man with a history of right parietal intracerebral hemorrhage 3 years ago, and multiple transient ischemic attacks, presented with confusion and bilateral temporo-occipital headache associated with nausea and multiple episodes of vomiting within 24 hours of his admission to the emergency department (ED). There was no history of hypertension, fall, recent sickness or travel. He was not on anti-platelet or anticoagulation therapy.
Upon physical examination, vital signs were normal, except a mildly elevated blood pressure (157/82 mm Hg). The patient was alert and oriented to time, place and person. He had a left visual field defect and sensory neglect. There was no gaze preference, aphasia or any focal motor deficit.
A brain CT scan in the ED showed large right parieto-temporal hematoma (Figure 1A). The patient was admitted to the stroke unit and was closely monitored. His blood pressure was treated to a level of ≤ 140 mmhg. Phenytoin was administered to prevent seizure spells. We performed magnetic resonance imaging (MRI) and conventional cerebral angiography on the patient to rule out underlying Arteriovenous malformation, dural arteriovenous fistula and other structural causes of brain bleeding. The cerebral angiography was unremarkable. However, the MRI brain showed surrounding edema out of proportion to the size of the hematoma.
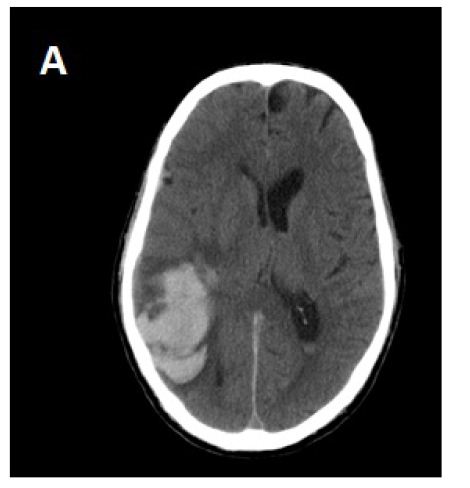
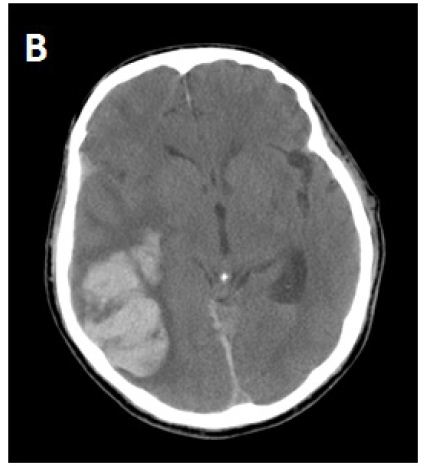
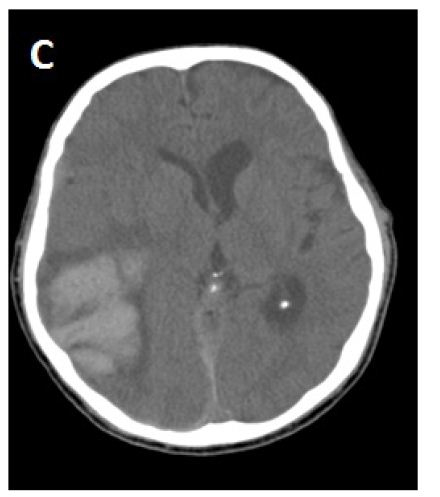
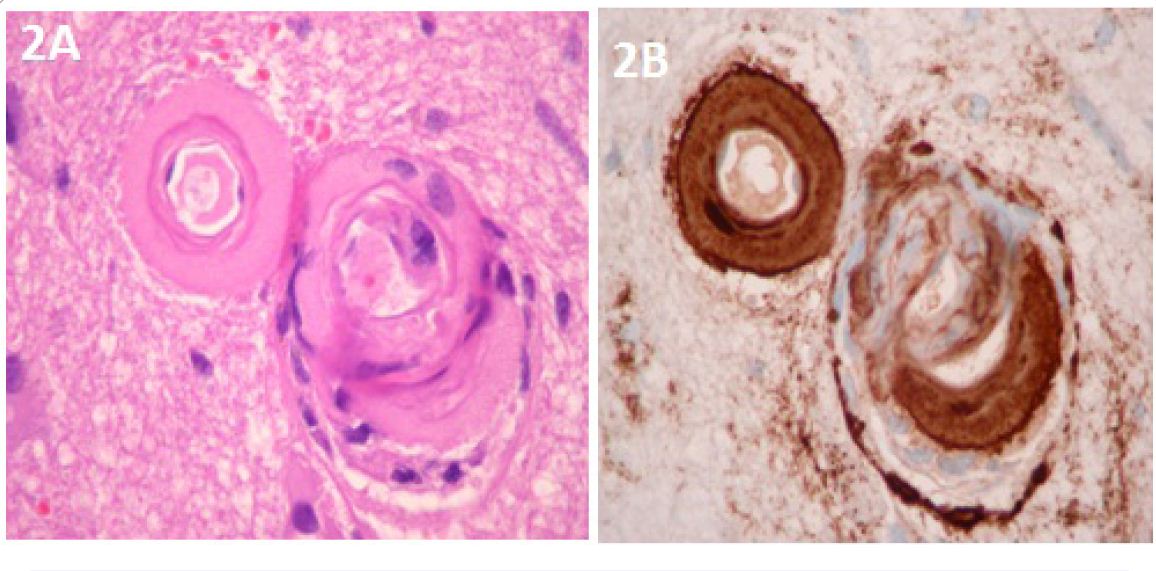
During the next several days, the patient clinically deteriorated and became more confused, lethargic, less responsive to verbal requests and developed left hemiparesis. A repeat CT brain study showed expansion of the right parietal hematoma with increasing edema and mid line shift (Figure 1B). The edema extended out to subcortical U-fibers. He was treated with 3% saline (goal sodium level between 145-150 mEq/L) and methyl prednisolone 150 mg IV every 8 hours. The patient clinically improved within the next 48 hours. There was some radiological improvement as the edema and midline shift decreased from the prior CT brain study without any significant change in hematoma size (Figure 1C). Steroid treatment was stopped after 3 days, and 3% saline was continued. The patient again deteriorated clinically and repeat CT brain again showed worsening of edema and midline shift following discontinuation of steroid therapy. The patient was started on mannitol, underwent emergent intubation and, then had a craniotomy to evacuate the hematoma. Brain tissue samples were taken for histopathological diagnosis. His condition did not improve after hematoma evaculation, and brain CT scan showed resolution of the right parietal hematoma, but a new left parietal hematoma had formed. The patient was extubated per family wishes, “comfort care” was administered, and he died a few hours later. The family agreed to a limited autopsy (brain only). There was deposition of amyloid in the walls of cortical blood vessels and white matter without any significant typical Alzheimer’s disease plaques in the cortical tissue (Figure 2). There was infiltration of cortical vessels with macrophages, which was thought to be a reaction to the brain bleed without any clear evidence of angiitis.
Our case is an example of CAA with lobar intracerebral hemorrhage and surrounding edema that is out of proportion to the size of the hematoma and is responsive to steroid therapy. In clinical practice, the clinical and radiological findings are atypical for a case of intracranial bleed due to amyloid angiopathy without inflammation as there was no early expansion of intracerebral hematoma, yet the patient clinically deteriorated over the course of 2 weeks. Patients with intraparenchymal hemorrhage typically worsen clinically in the 24-36 hours after the ictus. Furthermore, our patient responded to steroid therapy, and then his condition deteriorated when steroid therapy was discontinued. Modern management guidelines do not recommend routine use of steroids for intraparenchymal hemorrhage, and thus, steroid administration is no longer a common practice when there is brain hemorrhage. The radiological findings in this case were suggestive of vasogenic edema which served as rationale for a short course of steroid therapy. Vasogenic edema of the magnitude in this case was most likely due to underlying vasculitis which has been described in cases of CAA [10,11]. However, there was no evidence of vasculitis on brain autopsy. It is possible the vasculitis was missed as high dose methyl prednisolone was administered and may have ablated such findings, or it was overlooked in the brain necropsy sampling procedure.
Another interesting radiological feature of our case was the interval worsening of white matter brain lesions (WML) on the brain MRI scan 3 years after our patient had his first intracerebral bleed in the right parietal region. WML, a common finding in CAA, are clinically relevant but often neglected in the differential diagnosis, and may be associated with intracerebral hemorrhage [12]. CAA with WML has also been associated with vasulitis and the progression of these WML might serve as a marker of CAA [12].
More recently, a consensus group classified amyloid-related imaging abnormalities (ARIA) into those corresponding to edema or sulcal effacement (ARIA-E) and those associated with cerebral microbleeds or small hemorrhages (ARIA-H) [13,14]. As such, amyloid beta might exit via brain blood vessels due to an increase in vascular permeability. This may lead to leakage of proteinaceous substances in ARIA-E, and blood products in ARIA-H [15,16]. Furthermore, an aggressive form of CAA called CAA-ri, as we speculate occurred in our case, is characterized by vascular or perivascular inflammation. Our patient diplayed typical manifestations of CAA-ri which may include rapid and progressive cognitive decline, focal neurological deficits, and seizure. Like ARIA, CAA-ri typically resolves spontaneously or with steroid therapy. Recently, it has been shown that in CAA-ri patients, cerebrospinal fluid amyloid beta antibodies increase with symptomatic CAA-ri, but decrease with clinical and neuroimaging remission of CAA-ri, and are not elevated in sporadic CAA without inflammation [17]. Administration of oral corticosteroids has been shown to significantly decrease levels of anti- Aβ 42 antibody in CSF along with improvement in clinical symptoms [6]. This biomarker may prove important in future management of patients such as our patient who may have had CAA-ri.
In summary, we provided the clinical findings and course of disease of an elderly patient with a past history of lobar brain hemorrhage and recent history of recurrent lobar brain hemorrhages. Brain imaging showed evidence of past cerebral microbleeds and extension of edema to the U-fibers surrounding the acute brain hemorrhage. In addition, the patients’ edema was out of proportion to the size of hematoma. In such clinical circumstances, one may consider underlying vasculitis or CAA-ri, and in these cases steroids or other immunotherapy may be of benefit.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
