
Case report | DOI: https://doi.org/10.31579/2578-8868/323
*Corresponding Author: Anam Tariq, Department of Surgery, Aga Khan University Hospital, Karachi, Pakistan.
Citation: Anam Tariq, (2024), Central Precocious Puberty in a 5-year-old Boy with Hypothalamic Hamartoma: A Case Report and Review of the Literature, J. Neuroscience and Neurological Surgery, 15(5); DOI:10.31579/2578-8868/323
Copyright: ©, 2024 Anam Tariq. This is an open-access article distributed under the terms of The Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Received: 03 June 2024 | Accepted: 20 June 2024 | Published: 04 July 2024
Keywords: Hypothalamic hamartoma, precocious puberty, Gonadotropin-releasing hormone
Background: Central precocious puberty (CPP) occurs due to the precipitous production of gonadotropin-releasing hormone (GnRH) and premature activation of the hypothalamic-gonadal axis. It is the most typical endocrine symptom of hypothalamic hamartoma. Hypothalamic hamartomas (HH) are rare, non-cancerous malformations of the central nervous system (CNS). They can show up as CPP related to the release of luteinizing hormone in addition to developmental delay, visual impairments, hyperactivity, or gelastic seizures in young children.
Case Description: In this study, we present the case of a five-year-old Afghan child who displayed quickly evolving secondary sexual traits linked to hypothalamic dysfunction. Endocrine testing supported the central underlying cause of early puberty. The early detection of a small-sized, rounded lesion at the base of the tuber-cinereum on Magnetic Resonance Imaging (MRI) led to the diagnosis of Hypothalamic hamartoma (HH). To delay puberty, the patient was treated conservatively with a long-acting gonadotropin-releasing hormone (GnRH) analogue and his response was monitored.
Conclusion: This case report includes a review of the relevant literature as well as possible management strategies for a hypothalamic hamartoma.
Precocious puberty occurs when the first signs of puberty appear before the age of 8 for girls and 9 for boys. Early activation of the hypothalamic-pituitary-gonadal axis causes central precocious puberty and has several potential causes, including congenital abnormalities, radiation, ischemia, hydrocephalus, and infections; however, an organic lesion must be ruled out when it occurs in children [1].
Hypothalamic hamartoma, the most frequent cause of CPP, is a rare heterotrophic CNS malformation made up of disorderly organized neural tissue. Hypothalamic hamartoma is the aetiology of 75% of occurrences of CPP in children. Based on anatomy, the two main types of hypothalamic hamartomas—sessile and pedunculate—are commonly distinguished. They manifest as distinct spherical masses. However, mixed types make up more than 45% of HH [1,2].
These frequently manifest in the first decade of life as cognitive impairment, gelastic (laughing) seizures, and behavioral problems, along with clinical features of precocious puberty [1,2]. The most critical imaging technique for determining the presence of a hypothalamic hamartoma is magnetic resonance imaging (MRI) [2,3]. Medical management is typically effective in treating precocious puberty. Leuprolide acetate prevents the release of GnRH, which is necessary to initiate puberty. However, surgical resection is the gold standard of treatment [3].
In this case report, we address the case of a five-year-old boy who developed CPP because of a hypothalamic hamartoma, along with the early onset of pubic hair development, penile enlargement, and a significant rise in growth rate. His hormonal profile matched the CPP and the puberty was in line with Tanner's stage 4. His psychomotor capabilities were entirely normal, and he had no history of seizures. He was treated with a gonadotropin-releasing hormone (GnRH) agonist, and a follow-up appointment was planned for three months later to determine the status of his puberty.
A five-year-old boy from Afghanistan visited the neurosurgery clinic with signs of precocious puberty, including a rapid growth spurt, penile enlargement, and excessive pubic and axillary hair growth. He also had symptoms of hypothalamic dysfunction, including behavioral problems, urinary incontinence, and increased appetite. His birth history was unremarkable, and he had no history of developmental delay, birth abnormalities, or gelastic seizures.
At the time of his arrival, he was a tall child, standing 131.5 cm tall and weighing 33 kg. Physical examination indicated breast Tanner stage II, pubic hair Tanner stage III, and growth Tanner stage IV. The rest, as well as the neurological exam, was completely unremarkable. His laboratory workup [ Table 1] showed high testosterone levels of 7.57 ng/ml, luteinizing hormone (LH) levels of 5.5microIU/ml, and follicle-stimulating hormone (FSH) levels of 44microIU/ml, respectively. These results were compatible with the central cause of his precocious puberty. The levels of prolactin (12ng/ml), thyroid stimulating hormone (3.3microIU/ml), cortisol (10mcg/dL), and free T4 (1.35ng/dl) were all within normal ranges.
| HORMONE | MEASURED VALUE | NORMAL RANGE |
|---|---|---|
| TESTOSTERONE | 7.57 | 0.1-5.0 ng/ml |
| LH (LEUTINIZING HORMONE) | 5.5 | 2.0-10.0 mIU/ml |
| FSH (FOLLICLE STIMULATING HORMONE) | 44 | 3.0-9.0 mIU/ml |
| PROLACTIN | 12 | 0.0-20.0 ng/ml |
| TSH (THYROID STIMULATING HORMONE) | 3.3 | 0.2-4.7 mIU/ml |
| CORTISOL | 10 | 5.0-25.0 mcg/dl |
| FT4 (FREE TETRAIODOTHYRONINE) | 1.35 | 0.7-1.9 ng/dl |
Table 1: Laboratory Work-Up
To rule out the origin of the central precocious puberty, brain magnetic resonance imaging (MRI) was performed. T1-weighted imaging revealed a pedunculated, well-defined, non-contrast enhancing iso-dense mass in the tuber-cinereum that was compatible with hypothalamic hamartoma [Figure 1]
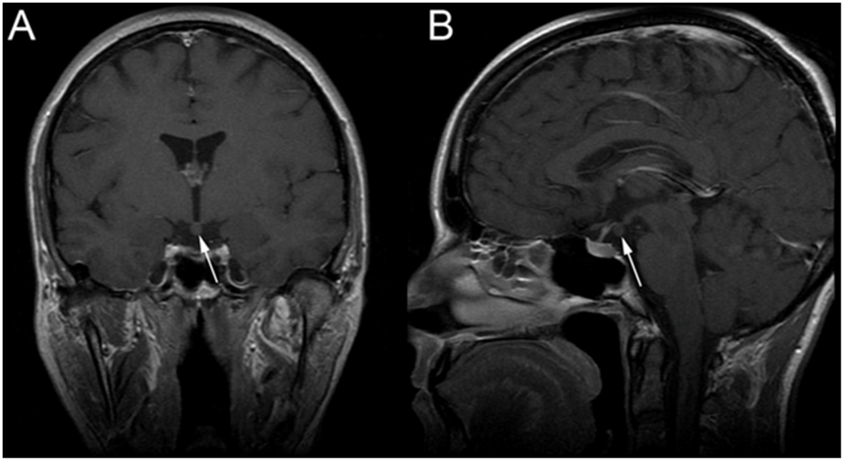
Figure 1: Magnetic Resonance Imaging (MRI) A hypothalamic lesion (arrows) is visible on the coronal (A) and sagittal (B) contrast-enhanced T1-weighted sequences, emerging from the third ventricle's floor. It is isointense to grey matter and is non-contrast enhancing suggesting hypothalamic hamartoma.
After receiving a diagnosis, the patient was started on the hormonal treatment with GnRH analogues; INJECTION LUPRON 7.5mg intramuscularly every four weeks. Surgery was deferred due to the modest size of the lesion. To track the development of his puberty, the patient was advised to have three-monthly follow-ups. Eventually, he had remarkable improvement in his symptoms.
Hypothalamic hamartomas are a development from the third ventricle's floor and are found at the base of the hypothalamus. These are uncommon, non-progressive, congenital, CNS-isolated lesions that are not cancerous. Most frequently, they manifest as CPP or the early beginning of puberty in young children. In addition, they can manifest as behavioral abnormalities, cognitive deterioration, and gelastic seizures. Although the prevalence of hypothalamic hamartoma is unknown, it is unquestionably the most frequent organic cause of CPP in young children, particularly in boys, with the female-to-male ratio being 1.3 to 1[1,3]. They frequently appear as independent lesions but up to 5% of cases are linked to Pallister-Hall syndrome [2,3].
The clinical features often correspond to the type of lesion. Infiltrative lesions typically appear as seizures, but pedunculated lesions are more frequently linked to CPP. The first signs can appear as early as birth. These include short stature, breast development, pubic hair, maturation of the sexual reproductive organs, voice deepening, and acne [3]. Due to his isolated CPP when he first appeared, our patient was thought to have a pedunculated lesion. A small non-enhancing lesion that is often iso-dense to the brain can be seen on a head computed tomography scan (CT scan) close to the interpeduncular and suprasellar cisterns. However, this imaging technology is now regarded as insufficient. Although electroencephalography (EEG) may be helpful when other seizure types are present, it typically appears normal during gelastic seizures. The gold standard for diagnosis is a brain MRI and the absence of contrast enhancement on MRI is crucial since it helps to identify the tumor from less prevalent regional diseases including hypothalamic astrocytomas, germ cell tumors, and Langerhans cell histiocytosis [2,3].
Hormonal therapy is typically advised for hypothalamic hamartoma that presents with CPP with surgery being reserved for refractory cases [3]. In their study, Kotwal, et al. [4] described a case of a 16-month-old girl who presented with fast-progressing sexual features at six months of age and vaginal hemorrhage. A well-defined, sessile hypothalamic mass was suggestive of a hypothalamic hamartoma discovered on magnetic resonance imaging (MRI) of the brain. On a two-year follow-up, she responded favorably to monthly GnRH analogue (Triptorelin) injections. Amin, et al. [5] reported a case of an 11-year-old boy who displayed early development of secondary sexual characteristics and whose MRI revealed a pedunculated hypothalamic hamartoma. Leuprolide acetate, a gonadotropin-releasing hormone (GnRH) analogue, was used to manage him conservatively, and a six-month follow-up revealed that his puberty had been arrested. Suh, et al. [6] similarly concluded that GnRH agonist therapy is both safe and efficient in treating CPP caused by HH.
Surgery is typically needed for patients who have uncontrolled seizures or have associated behavioral and psychiatric symptoms. Resection using a craniotomy has been the most widely used form of therapy. Transcallosal inter-forniceal resection, pterional resection, transtemporal trans choroidal resection, or combination resection for severe lesions are some of the methods that can be performed. The use of radiosurgery with a gamma knife, along with stereotactic thermoablation, to treat the lesion has also been documented in the literature. According to Castinetti, et al. [7], gamma knife treatment for hypothalamic hamartoma is a safe and efficient procedure. Agrawal, et al. [8] conducted a systematic review to determine the extent to which surgery is used to treat hypothalamic hamartoma (HH) which causes isolated central precocious puberty (CPP). The study showed that 73.6% of patients with pedunculated HH were cured, while 17.1% had partial relief. Only 11.1% of patients with sessile HH were cured. Of all patients with a pedunculated hamartoma who underwent total resection 88.88% were cured of the symptoms. Surgery had no effect in 73.9% of patients with sessile HH who underwent partial excision. The study concluded that surgical resection may be appropriate for some patients, particularly for tiny pedunculated hamartomas. A multidisciplinary strategy comprising neuro physicians, neurosurgeons, and endocrinologists produces the best results, according to research by Doddamani, et al. [9]. According to Alomari, et al. [10] the neurosurgeon should be adaptable when choosing a treatment strategy for a specific patient with HH and should be knowledgeable about the benefits and drawbacks of each choice. The severity of the clinical manifestations, the length and rate of progression of the clinical condition, the anatomy, location, and size of the HH, as well as the patient's age, all contribute to the choice of treatment.
There are some limitations to our study, which should be noted. Since this was a case report, it could not exhibit broad knowledge. Furthermore, the lack of biopsy prevented a conclusive diagnosis from being made. In conclusion, the best way to treat isolated CPP is under an endocrinologist's supervision and medical management with GnRH analogues is the mainstay of treatment. The severity of the clinical manifestations, the length and rate of progression of the clinical condition, the anatomy, location, and size of the HH, as well as the patient's age, all contribute to the choice of treatment. In all situations of drug-resistant epilepsy, early seizure control should be the aim, and patients should be quickly referred for surgery.
None.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
