
Case Report | DOI: https://doi.org/10.31579/2690-8794/084
1 Department of Medicine, Melaka Manipal Medical College, Melaka 75150, Malaysia.
2 Oncohematologist, Cancer Center of Guam, Tamuning, GU 96913, USA
*Corresponding Author: Mra Aye, Department of Medicine, Melaka Manipal Medical College, Melaka 75150, Malaysia.
Citation: Aye M., Cabot J. (2021) Case report of a Rare Cause of Portal Hypertension. Clinical Medical Reviews and Reports. 3(6); DOI: 10.31579/2690-8794/084
Copyright: © 2021, Mra Aye, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 03 May 2021 | Accepted: 19 June 2021 | Published: 13 August 2021
Keywords: william-beuren syndrome, portal hypertension
Williams or Williams-Beuren (WBS) is a developmental disorder with multisystemic manifestations. Chromosome 7 microdeletion underlying WBS occurs because of the unique genetic architecture in this region. Facial features change from subtle to dramatic. The extent of mental and development problems is variable. Cardiovascular, endocrine, and nervous system involvement mostly affect the morbidity and mortality. Although many systems are involved in this syndrome, portal hypertension and splenomegaly are scarcely reported. We report a case of William syndrome with moderate splenomegaly and portal hypertension.
Williams or Williams-Beuren syndrome (WBS) syndrome is estimated to occur in approximately 1 in 10,000 persons. [1] It is characterized by distinctive facial features, mental disability with unique personality features. It is caused by deletion of the WBS chromosome region, spanning 1.5 million to 1.8 million base pairs and containing 26 to 28 genes [1]. Exactly how gene loss leads to the characteristic phenotype of WBS is unknown, but hypo expression of gene products is likely to be involved. It is caused by haploinsufficiency for genes deleted in chromosome band 7q11.23[1]. However, apart from arterial stenoses caused by haploinsufficiency for the elastin gene (ELN) no specific implication of any other gene in the phenotype has been established reported by Miguel Del Campo Casanelles et al [2].
Recognition of WBS syndrome usually starts with the astute clinician. Clinical diagnostic criteria [3,4] have only modest usefulness as compared with rapid and accurate laboratory testing. Fluorescence in situ hybridization (FISH) involving ELN-specific probes establishes the diagnosis of WBS by showing the presence of a single ELN allele only rather than two alleles. Multisystem manifestations such as development and cognition, auditory and ear, nose, and throat, cardiovascular, dental, endocrine, gastrointestinal, genitourinary musculoskeletal, neurologic, ophthalmologic, skin and integument, personality, behavior, and emotional well-being, skin, and integument and miscellaneous are reported. Widespread gastrointestinal manifestation such as colic, difficulty feeding, textured-food intolerance constipation gastroesophageal reflux abdominal pain of unclear cause, diverticulitis can occur in young adults rectal prolapse and celiac disease was reported. However, hepatobiliary manifestations of splenomegaly, portal hypertension and rupture esophageal varices are not mentioned [1,5]. Miguel Del Campo Casanelles et al [2] reported two patients with portal hypertension leading to splenomegaly and pancytopenia carrying the common 1.5 Mb WBS deletion. They proposed this is an additional severe vascular complication of ELN deficiency and discussed the specific characteristics of the portal venous tract that could explain the impact of ELN deficiency in that venous territory.
Our aim is to report a case of WBS with portal hypertension which is so far very rare in the world literature.
A 21-year-old male was admitted for viral fever syndrome. He is a known case of William syndrome with manifestations of characteristic facial and dental features, poor cognitive function, hypertension, reflux esophagitis, portal hypertension complicated by esophageal varices and rupture and cryptogenic cirrhosis, adenotonsillar hypertrophy, obstructive sleep apnea, dental caries, and repeated otitis media. History by his mother reveals that he is the last child of her four children. Family history was unremarkable. Pregnancy and delivery at 42 weeks were uneventful. He had delayed milestones compared to her other children and was difficult to be fed. Initially he was diagnosed as Noonan’s syndrome during medical check- up when he went for hernia operation at age of 2 month. However, at age of 5 month a diagnosis of William syndrome was confirmed by genetic study. She was not sure whether FISH and molecular studies were carried out and she was unable to mention the result of genetic study result in detail. She mentioned that he had one episode of hypercalcemia in his infancy. At age of 2 he underwent emergency laparotomy when he developed abdominal pain and hematemesis, but postoperative diagnosis was intestinal obstruction.
Because of poor cognitive function, he needed to go to special school. He is dependent for all daily activities till the day of admission. At age of 5 he was diagnosed with severe esophageal reflux disease and young hypertension. However, they were informed that he has no heart disease. A few months later he developed hematemesis and melena and medical evaluation revealed ruptured esophageal varices and banding was done. Since then, he has had been doing regular check- up under hepatobiliary unit of at a tertiary hospital and ligation was done whenever it was necessary. The mother also mentioned that his platelet count is always lower than normal range. His last Esophago-Gastric-Duodenal-Scope was in Dec: 2018. He has repeated attacks gingivitis and dental caries, otitis media and nasal blockage due to Aden tonsillar hypertrophy with obstructive sleep apnea [but defaulted the investigations for obstructive sleep apnea, gingivitis secondary to bad dental caries and feeding disorder: he only eats semisolid food.
On examination, he was very friendly and cheerful. He had facial features of broad forehead, short nose and full cheeks and dental features of small or unusually shaped primary teeth and malocclusions, shown in [figure 1] consistent with William syndrome.
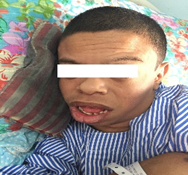
His height is 140cm. His limbs were shorter than his trunk. He walked, bathed, ate, and dressed in aids. His mental age was about age of 5 despite chronological age was 22. His BP was 145/85 mmHg. Systemic examination was normal [ no features of supravalvular aortic stenosis] except splenomegaly that extended 7 cm below the costal margin and very smooth skin texture. There was no clubbing of fingers, palmer erythema, spider nevi and gynecomastia and caput medusae, no ascites and edema of the legs.
Laboratory report. Full blood count: Hemoglobin 14.9g/dL, RBC 3.5x106/µL, WBC 3.5x103/µL, Platelet 42.6 x103/µL. BUSE: Sodium was 137, K 3.8, Chloride 106, HCO3 - 22 mmol/L. RP: urea is 36mmol/L and Creatinine is 63umol/L. Liver function test reveals total bilirubin of 33 umol/dl, total protein 66 where albumin is 38 and globulin is 28g/dL, Alkaline phosphate, Alanine amino aspartate and Alanine aminotransferase are 65, 81 and 43 IU/L respectively. Prothrombin time and APTT are 14 and 14.3 seconds respectively and INR is 1.24.

Echocardiogram report: Ejection fraction 74% No LVH. No RWMA, Dilated LA size, all valves morphology appeared normal, No clot, no pericardial effusion.
Ultrasonography report of liver and hepatobiliary system:
Liver is coarse in echotexture with slightly irregular margin. No focal liver lesions. Liver span measures 10.9cm. Gallbladder is well defined. No gallstone. Intrahepatic duct and common bile duct are not dilated. No intraduct calculus seen. Portal vein is patent (1.1cm). Pancreas is obscured by bowel gas. Spleen is enlarged but homogenous (18.7cm). No focal lesion. Both kidneys are normal in size and in echogenicity. BPL/PT right kidney: 9.4/1.cm: left kidney: 10.7cm/1cm No focal renal lesion or obvious calcifications. No renal calculus or hydronephrosis bilaterally. Urinary bladder is partially distended. No obvious bladder stone or mass
Impression: Liver cirrhosis with hepatosplenomegaly
This case is author’s first ever experience of William’s syndrome [1] with portal hypertension and rupture esophageal varices in 35 years as a general physician. Short stature, smooth skin, peculiar personality, poor cognitive functions, and poor dentition and associated with systemic involvement strike us a likely case of syndromic disease. However, we were unable to arrive at the diagnosis of WBS [1] unless we are informed by the mother. He has most of reported the features of WBS except its common feature of supravalvular aortic stenosis. Additionally, WBS with portal hypertension and splenomegaly and upper GI bleeding due to esophageal rupture is scarcely reported in the world literature. So far, two cases were reported by Miguel Del et al [2]. Those cases were reported to carry the common 1.5 Mb WBS deletion which were proposed to be associated with additional severe vascular complication of Elastin gene deficiency and discuss the specific characteristics of the portal venous tract that could explain the impact of Elastin gene deficiency in that venous territory.
Patients with WBS need lifelong medical attention as there are many manifestations with poor morbidity and mortality. A formal assessment of the life expectancy associated with WBS is lacking. One study of approximately 300 patients, 1 to 55 years of age, with WBS showed a cardiovascular-associated mortality 25 to 100 times that among the controls [6]. Cardiovascular manifestations are detected in about 75% of confirmed patients during their lifetime [7]. Supravalvular aortic stenosis (SVAS) is most common, peripheral pulmonic stenosis being next in frequency. Cardiovascular complications such as biventricular outflow obstruction, biventricular hypertrophy, or stenosis or occlusion of coronary ostia has been reported to increase the relative risk of adverse outcomes. Cardiovascular complications are the major cause of death in patients with WBS [8]. Vessel occlusions either arterial or venous territories are result of elastin gene deficiency [2]. Coronary artery occlusion and ventricular hypertrophy are risk factors for sudden deaths due to cardiac arrhythmias.
Fortunately, our patient has no cardiovascular involvement except hypertension and hypertensive heart disease. Hypertension is a manageable disease. However, our patient has portal hypertension and splenomegaly complicated by rupture esophageal varices. Nevertheless, he does not have signs of cirrhosis of liver both clinically and biochemically except thrombocytopenia and leucopenia in full blood picture and hyperechogenic liver in the ultrasound of liver. It can be explained by prehepatic of portal hypertension supporting the proposal by Miguel Del Campo Casanelleset et al [ 2] that portal hypertension may be an unusual complication in WBS related to the generalized dysplasia of the vascular wall of portal vein territory caused by elastin haploinsufficiency.
WBS with moderate splenomegaly complicated by pancytopenia and septic shock and death are also a major risk factors to shorten the lives of William syndrome [2] are scarcely reported which our patient has and therefore portal hypertension and splenomegaly should also be looked for in any patient diagnosed with WBS. Their two patients with portal hypertension were carrying the common 1.5 Mb WBS deletion. The great majority of patients show very similar 1.5 Mb heterozygous deletions that arise because of unequal crossing-over between highly homologous regional segmental duplications and include at least 25 identified genes [9-11].
While haploinsufficiency for the elastin gene is known to be the cause of the vascular stenoses, the other features of WBS have not been clearly attributed to specific genes and they proposed that this is an additional severe vascular complication of ELN deficiency [2].
Portal hypertension complicated by rupture esophageal varices, splenomegaly and hypersplenism are potentially lethal and should thus be looked for in any patient with WBS and splenomegaly.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
