
Mini Review | DOI: https://doi.org/10.31579/2640-1045/089
*Corresponding Author: Maurice Israël, 2 Av. Aristide Briand, 91190 Bures sur Yvette, France.
Citation: Israël M. (2021). Carcinogenic Krebs-Cycle Enzyme Mutations Supporting Ketolytic-Dependent Tumors. Endocrinology and Disorders. 5(6): DOI:10.31579/2640-1045/089
Copyright: © 2021 Maurice Israël, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 24 August 2021 | Accepted: 18 September 2021 | Published: 24 September 2021
Keywords: endocrine pancreas; SCOT; oxct1; carcinogenic mutations
Tumors incorporate much glucose for overcoming glycolytic pyruvate-kinase and pyruvate-dehydrogenase inhibitions; they form lactate, rather than oxidative acetyl-CoA. Tumors also need to synthetize fatty acids, which automatically turns-off their mitochondrial degradation into acetyl-CoA. Thus, ketolysis becomes their major acetyl-CoA supply. Carcinogenic mutations or deficiencies of Krebs-cycle enzymes support the ketolytic dependency of tumors.
In Cancer, Warburg showed an increased fermentation of glucose into lactic acid, even in presence of oxygen [1]. The explanation came from Eigenbrodt, who identified a poorly active pyruvate-kinase (PK) M2 isoform forming a “bottleneck” at the end of glycolysis, which then limited the entry in oxidative metabolism [2]. Moreover, there was also an inhibition of pyruvate- dehydrogenase (PDH) [3], which stops the conversion of pyruvate into acetyl-CoA and its entry in oxidative metabolism, orienting the glycolytic input toward lactic fermentation. These PK and PDH inhibitions by phosphorylation are usually associated to catabolic hormonal actions (glucagon, epinephrine and cortisol). Switching from catabolism, to anabolism mediated by insulin, requires a de-phosphorylation of PK and PDH; this process seems to be impaired in tumor cells. We earlier did the hypothesis that a deficient GABAergic regulation in the endocrine pancreas, would explain this situation [4]. Indeed, this regulation of the endocrine pancreas, normally turns-off catabolic glucagon release from alpha cells, while beta cells release insulin and GABA, the latter also puts an end to insulin release, via GABA auto-receptors of beta cells. A failure of this mechanism would incompletely turn-off insulin release, which would gradually desensitize insulin receptors of differentiated tissues, responding to catabolic glucagon, with a ketogenic and neoglucogenic metabolism. Only new stem cells, with new, not-desensitized insulin receptors, will receive the anabolic insulin message, in parallel to the glucagon signal from alpha cells. They will then keep PK and PDH in the “OFF” phosphorylated configuration, but display an anabolic response to insulin or IGF, able to support their mitotic anabolic demands. Differentiated cells are in this way programmed for providing nutrients, amino acid, glucose and ketone bodies, for the benefit of a population of stem cells that rewire their metabolic pathways in this special way, which gives them a metabolic advantage. In parallel, the anabolic message received by tumor cells increases the synthesis of fatty acids and lipids, for making new membranes for mitotic cells. The citrate efflux from mitochondria feeds the cytosolic fatty acid synthesis pathway, starting with ATP citrate lyase; then acetyl-CoA carboxylase (ACC) takes over, converting cytosolic acetyl-CoA into malonyl-CoA. These steps are crucial, because malonyl-CoA will turn-off the mitochondrial carnityl- tansporter of fatty acids, and interrupt their degradation by beta-oxidation into acétyl-CoA, if cells have to synthesize fatty acids for their mitosis. Hence, with the interruption of both the glycolytic and fatty acid supplies of acetyl-CoA in tumors, the major source for their mitochondrial acetyl- CoA becomes ketone bodies, formed in hepatocytes, responding to catabolic glucagon [5] (Figure 1).
Ketogenesis depends of four enzymes: 1- thiolase;
2- hydroxylmethylglutarylCoAsynthetase;
3- hydroxymethylglutarylCoAlyase;
4- betahydroxybutyratedehydrogenase, the latter will convert acetoacetate into betahydroxybutyrate. Released betahydroxybutyrate reaches tumor cells, and gives back acetyl-CoA by ketolysis in mitochondria. Three enzymes form the ketolytic pathway, (4 and 1) are the same as for ketogenesis (but work in the opposite direction). In between 4 and 1, the specific ketolytic enzyme is succinyl-CoA: 3-oxoacid-CoA transferase (SCOT). An inhibition of SCOT should then deprive tumor cell mitochondria of acetyl- CoA, since their glycolytic and fatty acid supplies of acetyl-CoA are not operational in tumor cells. Thus, SCOT (the oxct1 gene product) could well be their vulnerable point.
One substrate of SCOT is acetoacetate; it comes from betahydroxybutyrate, formed in liver by ketogenesis; supported by ketogenic diets. The other substrate succinyl-CoA, forms in the Krebs cycle, via alphaketoglutarate-dehydrogenase, fed by glutamine and glutamate, transaminated, in mitochondria.
We know several mutations or deficiencies of Krebs cycle enzymes; they support a carcinogenic metabolism by reinforcing the SCOT dependency of tumor cells (Figure 1).
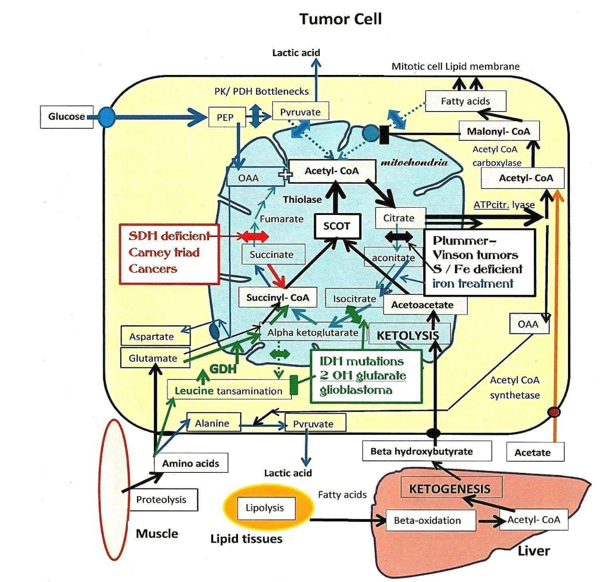
An increased glycolysis fails to overcome pyruvatekinase (PK) and pyruvatedehydrogenase (PDH) inhibitions. These bottlenecks close the glycolytic acetyl-CoA supply to mitochondria oxidative metabolism, explaining lactic- acid release. Since tumors have to synthesize fatty acid, malonyl- CoA, turns-off their mitochondrial transporter and degradation into acetyl-CoA. Thus, the mitochondrial acetyl-CoA supply will essentially come from ketolysis via succinyl-CoA: 3-oxoacid-CoA transferase (SCOT). This specific ketolytic enzyme transfers CoA from succinyl-CoA to acetoacetate provided by liver ketogenesis. Three Krebs cycle enzymatic deficiencies act on SCOT dependent tumores. In Plummer–Vinson dysphagia tumors, (black box interruption) mitochondrial aconitase devoid of its Iron-sulfur cluster (S/ Fe) is inactive, favoring the citrate efflux and the lipogenic pathway, needed for mitotic cells. On the contrary, when cytosolic aconitase receives an iron treatment, it becomes active, bringing back isocitrate in mitochondria, diverting it from lipogenesis. The next situation concerns the isocitrate dehydrogenase (IDH) mutations (green box interruption). These mutations found in glioblastoma form an abnormal oncometabolite (2 OH glutarate), it has epigenetic effects increasing branch-chain amino acid transaminase, but seems also to inhibit the transaminating activity, leucine will rise and boost Glutamate dehydrogenase ( GDH) feeding more efficiently SCOT with succinyl-CoA. The Carney triad tumors (Red box interruption) are Succinodehydrogenase (SDH) deficient mutations; succinate increases and feeds the synthesis of succinyl-CoA for SCOT. Note the exogenous acetate supply; the phosphoenol- pyruvate (PEP), oxaloacetate (OAA) entries in mitochondria.
First: tumors linked to iron and aconitase.
Aconitase activity requires a Sulfur/iron (S/Fe) cluster co-factor [6]. Without S/Fe, aconitate and isocitrate will not form, facilitating the mitochondrial citrate efflux, which then feeds fatty acid synthesis needed by mitotic cells. The inactive aconitase becomes an iron regulatory protein (IRP), which binds to iron responsive elements IREs on mRNAs, to control iron metabolism, and mitochondrial proteins that make the S/Fe clusters [7]. This complex machinery is made of a sulfur reductase, a scaffold protein, a chaperone and frataxin. The latter controlling the export of the S/Fe cluster towards various targets, such as mitochondrial aconitase, or cytosolic aconitase. An inhibition of aconitase, will increase the citrate efflux from mitochondria, and support the synthesis of fatty acids for mitotic tumor cells; this occurs if S/Fe is low or its export by frataxin deficient, as in Friedreich’s ataxia. On the other hand, an activation of cytosolic aconitase, should divert citrate from the fatty acid synthesis route, and bring it back in mitochondria. Indeed, in Plummer-Vinson dysphagia affecting women in Sweden, an iron treatment protected them from cancer (Gernez 1946) [8]. It is likely that cytosolic iron increases aconitase, diverting citrate away from lipogenesis, by bringing back isocitrate in the Krebs cycle.
Second: tumors associated to isocitrate dehydrogenase mutations (IDH1, IDH2).
These mutated enzymes associated to Gliomas, form an abnormal product, an oncometabolite:
D-2-hydroxyglutarate (2 OH Glut.). This compound has epigenetic effects increasing the expression of branched-chain amino acid-transaminases [9]. In spite of that, this oncometabolite seems to inhibit these transaminases [10]. Probably, leucine increases, and stimulates glutamate-dehydrogenase (GDH) [11] efficiently converting glutamate into alphaketoglutarate, which then increases the synthesis of succinyl-CoA the SCOT substrate, supporting the formation of tumor acetyl-CoA.
Third: tumors of Carney- Stratakis Syndrome
These succinodehydrogenase (SDH) deficient tumors [12] stop converting succinate into fumarate; thus, succinate gives back succinyl-CoA, via succinyl-CoA synthetase, boosting SCOT activity. This specific ketolytic enzyme forms acetoacetyl-CoA, and then acetyl-CoA via a thiolase, supporting again the supply of acetyl-CoA to tumor cells. These SDH- deficient tumors include gastrointestinal-intersticial tumors; pulmonary chondroma; paraganglioma, pheochromocytoma and renal carcinoma.
Note that (SDH) is the mitochondrial complex II, As for NADH dehydrogenase (Complex I), these flavoproteins have coenzyme Q for hydrogen acceptor. Increasing coenzyme Q would drag more succinate toward the electron-transport chain, leaving less substrate to SCOT. On the contrary, the GABA shunt involving glutamate decarboxylase (GAD) and GABAase, generates succinate and succinyl-CoA, for SCOT.
The carcinogenic mutations discussed show how tumors vitally depend of ketolysis for making mitochondrial acetyl-CoA; presumably, SCOT is a particularly vulnerable point. It could then be useful to inhibit SCOT and the enzymes that supply succinyl-CoA to SCOT, and to decrease the acetoacetate coming from an inappropriate high fat ketogenic diet. A possible effect of such diets is that butyrate a histone-deacetylase inhibitor, might decrease the oxct1 gene expression, but ketone bodies will also feed the keto-dependent tumors, which could be dangerous. After the citrate condensation step, citrate exits the mitochondria and gives back in the cytosol, acetyl-CoA via ATP-citrate-lyase, feeding the lipogenic pathway. There is also an acetyl-CoA coming from a cytosolic acetyl-CoA synthetase, incorporating directly exogenous acetate. It may be necessary to block this pathway, as previously done for ATP- citrate-lyase.
We are grateful to Guy Tenenbaum, and Eric Berg for a helpful discussion
The author declares no conflicts of interests.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
