
Research Article | DOI: https://doi.org/10.31579/2578-8965/220
Department of Obstetrics and Gynecology, Guizhou Provincial People’s Hospital.
*Corresponding Author: Yan Gao, Department of Obstetrics and Gynecology, Guizhou Provincial People’s Hospital.
Citation: Jie Zheng, Yan Gao, (2024), Bioinformatics-based analysis of EMT-related genes in cervical cancer patients, J. Obstetrics Gynecology and Reproductive Sciences, 8(7) DOI:10.31579/2578-8965/220
Copyright: © 2024, Yan Gao. This is an open-access article distributed under the terms of The Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 13 May 2024 | Accepted: 24 May 2024 | Published: 04 September 2024
Keywords: cervical cancer; bioinformatics analysis; EMT; differentially expressed genes; protein-protein interaction network; immune infiltrated cells
Background: In most cases, patients who are not screened canonically have advanced disease by the time they are first diagnosed. Therefore, invasive metastasis is the main feature and cause of death in patients with advanced cervical cancer.
Methods: The GEO database was searched for analytical data on cervical cancer. The GSE9750 dataset was employed to identify differentially expressed genes (DEGs) in tumor samples compared to normal samples, carry out enrichment analysis, build protein-protein interaction (PPI) networks to identify key genes (Hub gene), and perform between diagnostic genes and immune infiltrated cells.
Results: A total of 1440 DEGs and 46 differential genes linked to EMT were screened. GO enrichment produced 455 results, and Kyoto Encyclopedia of Genes and Genomes pathway enrichment (KEGG )yielded a total of 18 pathways. A PPI network was built using 46 differential genes related to EMT, and 9 Hub genes were also validated. Nine hub genes had exactly the same up- and down-regulation trend as before in the external verification set, but only six hub genes were ultimately determined to be diagnostic genes after further validation by the external verification set. The results of the KM (Kaplan-Meier) survival study also revealed five prognostic genes related to survival, namely MMP1, CXCL8, SSP1, MMP3, and VCAM1.
Conclusion: The genes MMP1, CXCL8, SSP1, MMP3, and VCAM1are crucial in the development of EMT in patients with cervical cancer and can be employed as both diagnostic and therapeutic targets during the progression of EMT in cervical cancer patients.
Cervical cancer (CC) is the second most common gynecological tumor and the leading cause of death in women1.Over 99% of precancerous lesions (cervical dysplasia) and cervical carcinomas are caused by high-risk HPV infection2 .A number of biomarkers [e.g. squamous cell carcinoma antigen (SCC-Ag)] are currently used in the diagnosis and prognosis of cervical cancer. However, the lack of sensitivity and specificity of these biomarkers limits their utility3.At present, the treatment of cervical cancer mainly includes surgery, radiotherapy, chemotherapy and immunotherapy4. In recent years, with the continuous development of medical technology and the application of cervical cancer vaccines, there are more accessible personnel and technologies to implement appropriate treatments, so the mortality rate of cervical cancer in developed countries has decreased5.However, for many
underdeveloped nations, the scarcity of resources and infrastructure make such preventative and treatment programs limited or even non-existent. Currently,>85% of cervical cancer deaths occur in low and middle-income countries. Tragically, cervical cancer is the leading cause of cancer deaths in women of the developing world 6.The development of cervical cancer is associated with various factors such as chromosomal aberrations, DNA mutations, abnormal methylation, and abnormal regulation of pathways 7-10. Therefore, in order to find a simpler and more reliable diagnosis method, explore the potential mechanism of related genes affecting cervical cancer cells, and reduce the mortality rate of cervical cancer, it is still necessary to further develop diagnostic markers and therapeutic targets for cervical cancer to provide new methods for the treatment and intervention of patients.
Cancer cells acquire metastatic potential and gain motility primarily through the EMT process. EMT is a cellular process that is characterized by a change from an epithelial to a metachromatic phenotype11. This process is crucial for cancer development, invasion, metastasis, tumor immunosuppression, and immune evasion12. Clinical data indicate that EMT is strongly linked to a poor prognosis in patients with cervical cancer13 . Several studies have now explored the mechanisms of cervical lymph node metastasis14,15 . However, there are still no validated biomarkers associated with metastasis during the progression of EMT in cervical cancer patients.
The above studies have shown that EMT has an important role in the progression of cervical cancer. Therefore, this study aims to find EMT-related genes that influence the development of cervical cancer.
2.1 Screening and differential expression analysis of DEGs
The cervical cancer analysis data were first extracted from the GEO database, and GSE9750 served as the training set. The DEGs were screened based on the GSE9750 dataset, and the limma package was used to analyze the DEGs and produce a volcano plot with the screening conditions of p.adj < 0>1.Heat maps of DEGs expression were plotted using the ggplot2 package in R software.
2.2 Acquisition of EMT-related differential genes and analysis of GO and KEGG enrichment
Venn diagrams were used to identify the intersection genes of DEGs with EMT-related genes, and GO functional annotations and KEGG enrichment analysis of EMT-related differential genes were performed using the R package (package cluster Profiler). One of the GO functional annotations includes biological processes, cellular composition and molecular functional analysis to predict the function of the protein. KEGG enrichment analysis will concentrate DEGs in specific pathways to construct a network of intermolecular interactions and relationships. Parameters are set to default (adj. p value < 0>
2.3 Screening of key HUB genes and ROC curve test
Import EMT-related differential genes into STRING database to construct protein interaction network (default parameters). Then, the analysis results were imported into Cytoscape software and the Hub gene was screened using the plug-in MCODE (default parameters: degree cutoff:2;Node score cutoff:0.2;K-core:2;Max.depth:100). The PPI screened Hub genes were submitted to expression and ROC analysis. Box-line plots of Hub gene expression were drawn; ROC analysis was subsequently performed.
2.4 External validation of HUB genes
The expression and ROC of 9 Hub genes were validated in the external validation set GSE127265, which included 10 samples, including 7 tumor samples and 3 normal samples. The expression of the nine Hub genes in the control and CC groups were compared, respectively, to observe whether the up- and down-regulation relationships of each Hub gene were consistent with those in the previous step 2.3, and to observe whether the AUC values in the ROC analysis were greater than 0.7.
2.5 Correlation analysis of diagnostic genes
Correlations between the six diagnostic genes were calculated based on spearman correlation analysis using the R package.
2.6 Infiltrating cell correlation analysis of diagnostic genes
In this study, immune infiltrating cell analysis was performed on the sample gene expression data using the R package CIBERSORT algorithm to calculate the sample immune infiltrating cell abundance. The correlation coefficient (r-value) and significance (p-value) between differential immune infiltrating cells and diagnostic genes were calculated using R package psych based on the sample immune infiltrating cell abundance and 6 diagnostic gene expression matrices. The R package ggpubr was used to create a lollipop plot of each hub gene associated with immune infiltrating cells.
2.7 Mapping the gene network
The regulatory mechanisms among diagnostic genes, TFs, and miRNAs were analyzed by the online miRNet (https://www.mirnet.ca) database to derive the linkage between Gene - TFs and also between Gene - miRNAs.
3.1 Dentification of differential genes
Based on the GSE9750 dataset, 1440 DEGs were screened between cervical cancer samples and normal samples, including 415 differentially up-regulated genes and 1025 differentially down-regulated genes (Figure 1); The most significant 10 up- and down-regulated genes each in Top logFC were selected for heat map (Figure 2)
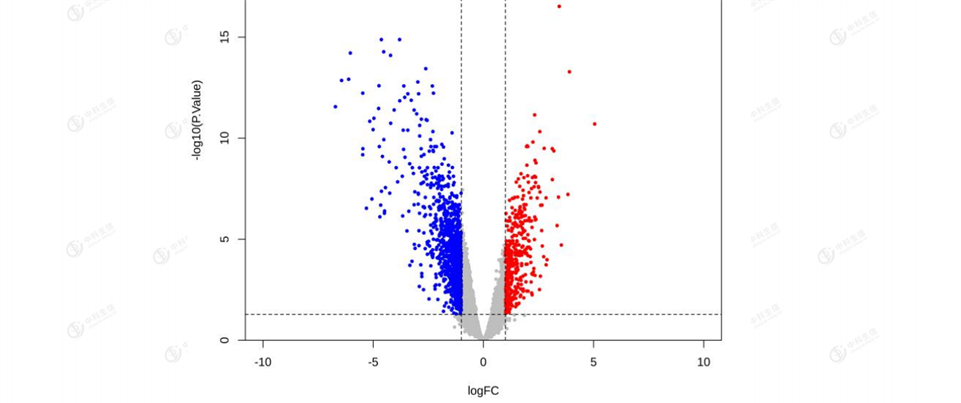
Figure 1: Volcano plot of differentially expressed genes. The vertical coordinates indicate the corrected P-value (the logarithm of the base 10 is taken for the corrected P-value to make the image more aesthetically pleasing); the horizontal coordinates indicate logFC (Fold Chang). Grey:non-significant genes; red : Red: up-regulation;blue:down-regulation.
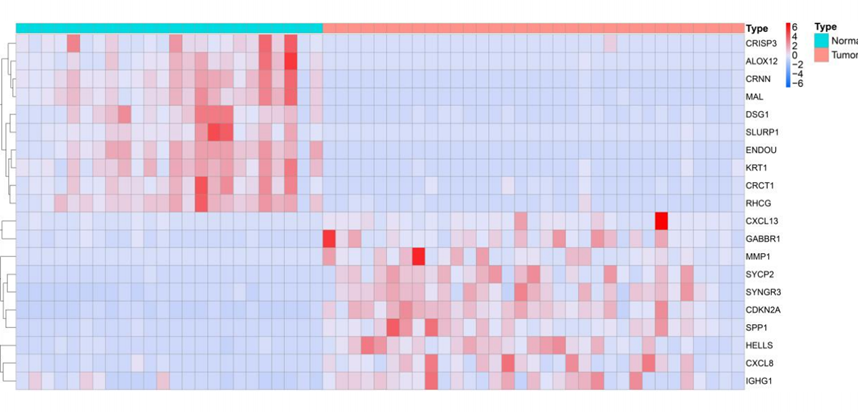
Figure 2: Heat map of differentially expressed genes.
3.2 Identification of EMT-related genes and GO and KEGG enrichment analysis
Direct intersection of DEGs with EMT-related genes resulted in 46 intersected genes (Figure3). 455 results were obtained from GO enrichment, of which 417 BPs (biological processes), 32 MFs (molecular functions), and 6 CCs (cellular components) were enriched. The results showed that in BP, the shared differential genes were mainly enriched in "cell growth regulation" and "extracellular matrix". In CC, shared differential genes are mainly enriched in "collagen-containing extracellular matrix" and
"endoplasmic reticulum lumen"; in MF, shared differential genes are mainly enriched in "structural components of extracellular matrix" and "integrin binding" molecular functions (Figure 4). These results suggest that immune and metabolic changes are necessary for tumorigenesis and progression. KEGG is enriched in a total of 18 pathways, and the differential genes are mainly enriched in "TNF", "Wnt", "IL-17" and other signaling pathways. "This indicates that most of these target genes are enriched in cancer-related pathways, and one gene may be involved in multiple pathways (Figure 5).
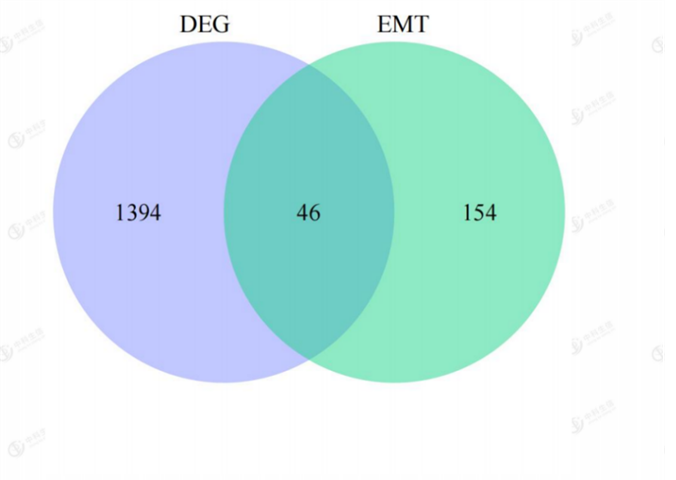
Figure 3: Venn diagram of the intersection of EMT-related differential genes;Purple:differentially expressed genes(DEG);Green:EMT-related genes;Bule:EMT-related differential genes.
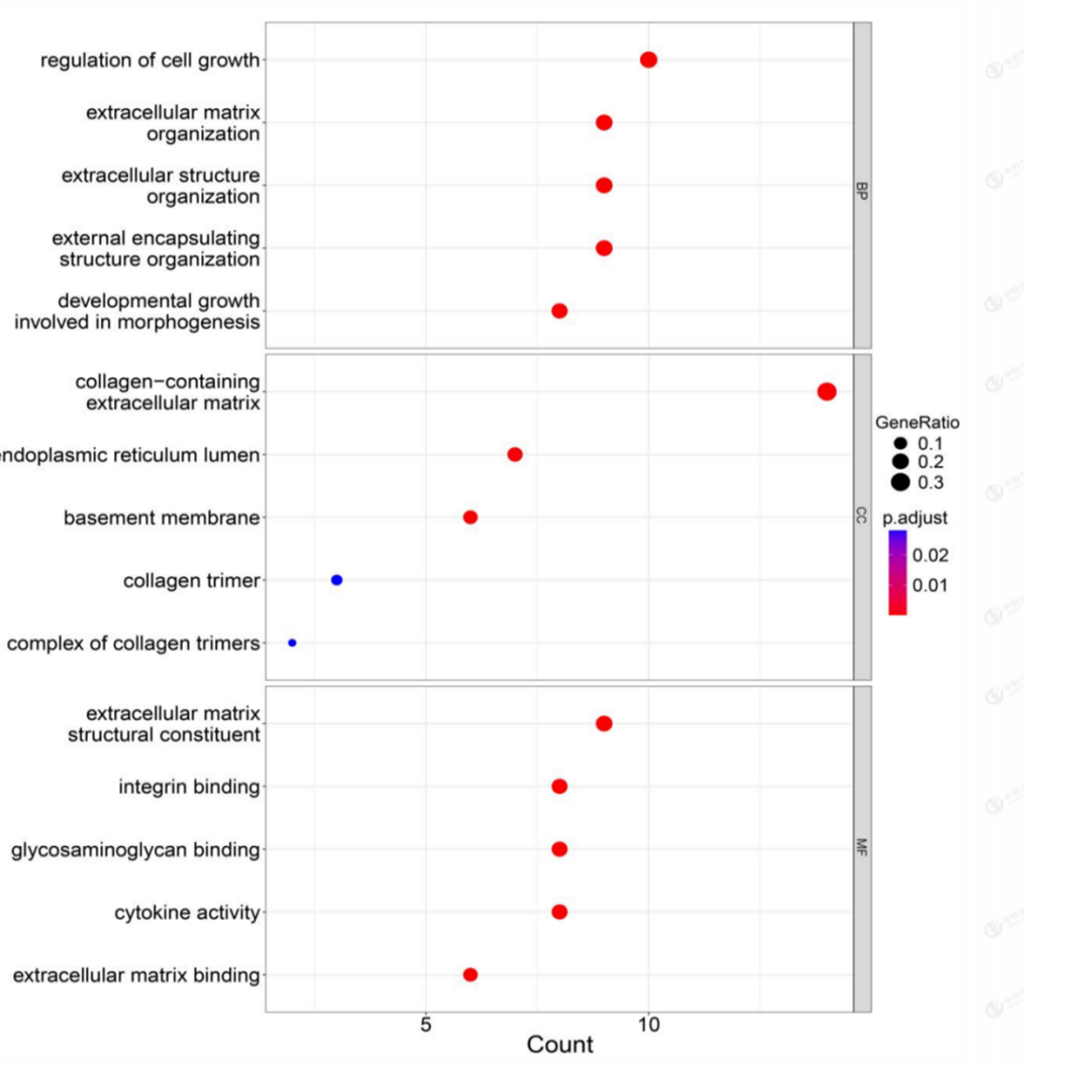
Figure 4: GO terms enrichment analysis of DEGs.
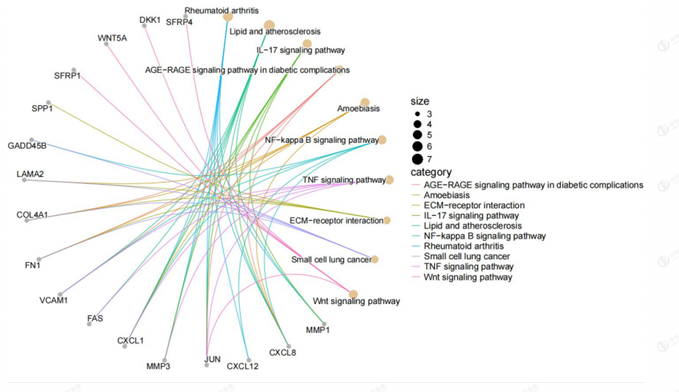
Figure 5: Relation graph of KEGG pathways in which differentially expressed genes were significantly enriched. There were 10 pathways in the relation graph.
3.3 Construction of PPI network to screen key Hub genes
A shared differential gene PPI network graph consisting of 46 EMT-related differential genes was constructed using the STRING online database, and 9 Hub genes were screened by applying the plug-in MCODE plug-in (Figure 6). The nine Hub genes are FN1, MMP1, CXCL8, SPP1, JUN, MMP3, CXCL12, CXCL1, and VCAM1( Figure 7 and Figure 8).
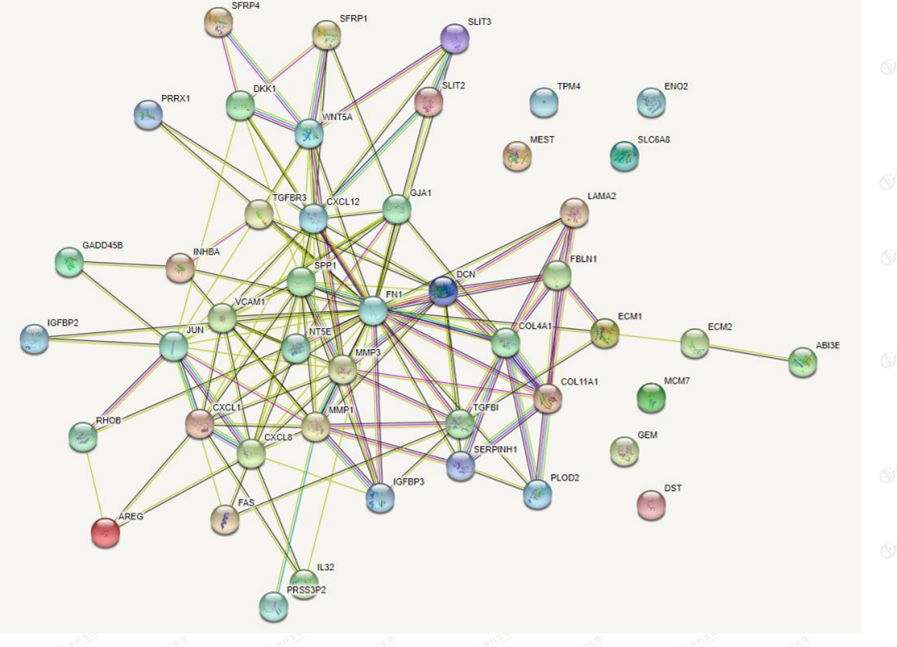
Figure 6: protein-protein interaction network.
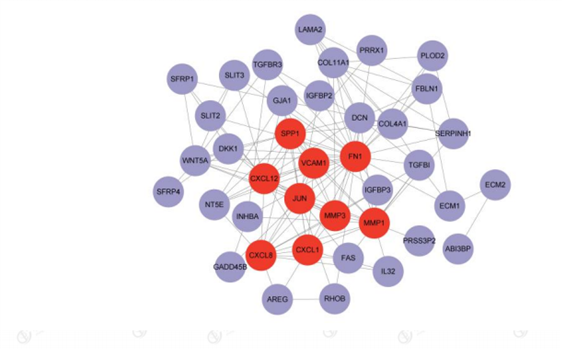
Figure 7: Total Gene Interaction Network.Red:9 Hub genes;Purple:The remaining genes in the 46 EMT-associated differential genes.
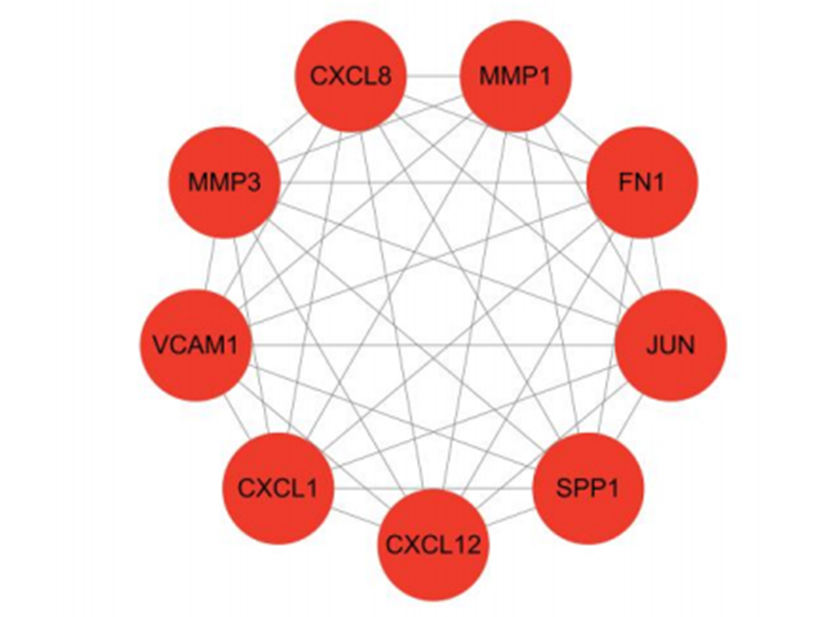
Figure 8: MCODE Screening Core Gene Network
3.4 Hub gene expression and ROC analysis
The box line plot of Hub gene expression was drawn based on the nine Hub genes screened by PPI for [removed]Figure 9), and the results showed that
among the nine Hub genes, JUN and CXCL12 showed down-regulation, while the rest were all in up-regulation relationship. ROC analysis was also performed (Figure 10), and the AUC values were all greater than 0.7.
Figure 9: Hub gene expression analysis.
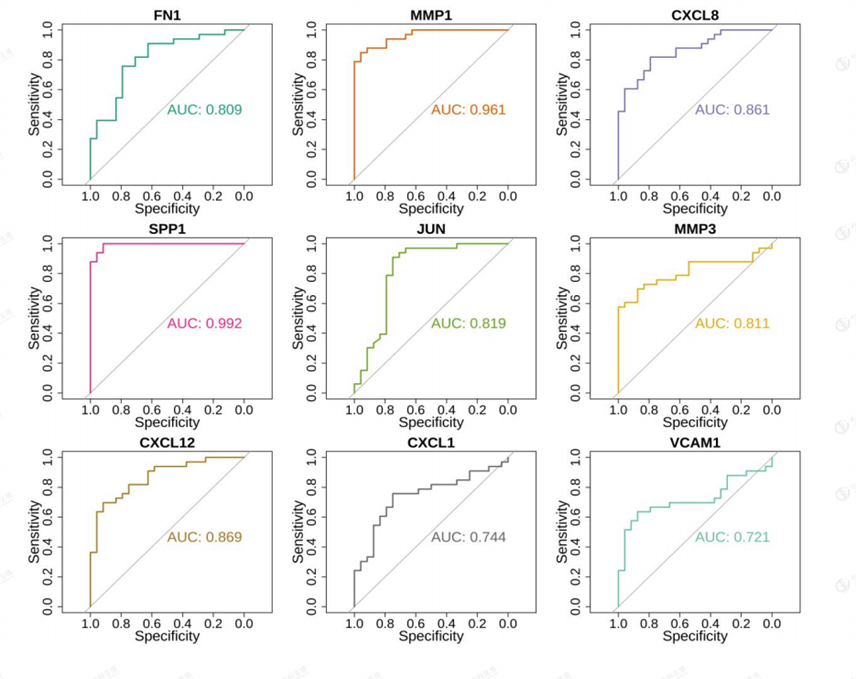
Figure 10: Hub gene ROC analysis.
3.5 External Validation Set Validity
The results showed that the differential expression of nine Hub genes in the external validation set was completely consistent with the previous one, but the p.adj values of three of them, FN1, JUN, and CXCL1, were not significant at greater than 0.05. ROC analysis showed that all AUC were greater than 0.7 (Figure 12). We selected diagnostic genes those genes whose up- and down-regulation relationships obtained from expression in the
external validation set were consistent and significant with the expression in 3.4, while meeting an AUC value greater than 0.7. The diagnostic genes meeting the above criteria were MMP1, CXCL8, SPP1, MMP3, CXCL12, and VCAM1.That is, the above six genes can be considered as genes with specificity in the progression of EMT in cervical cancer patients and can be used as markers for the diagnosis of their progression.
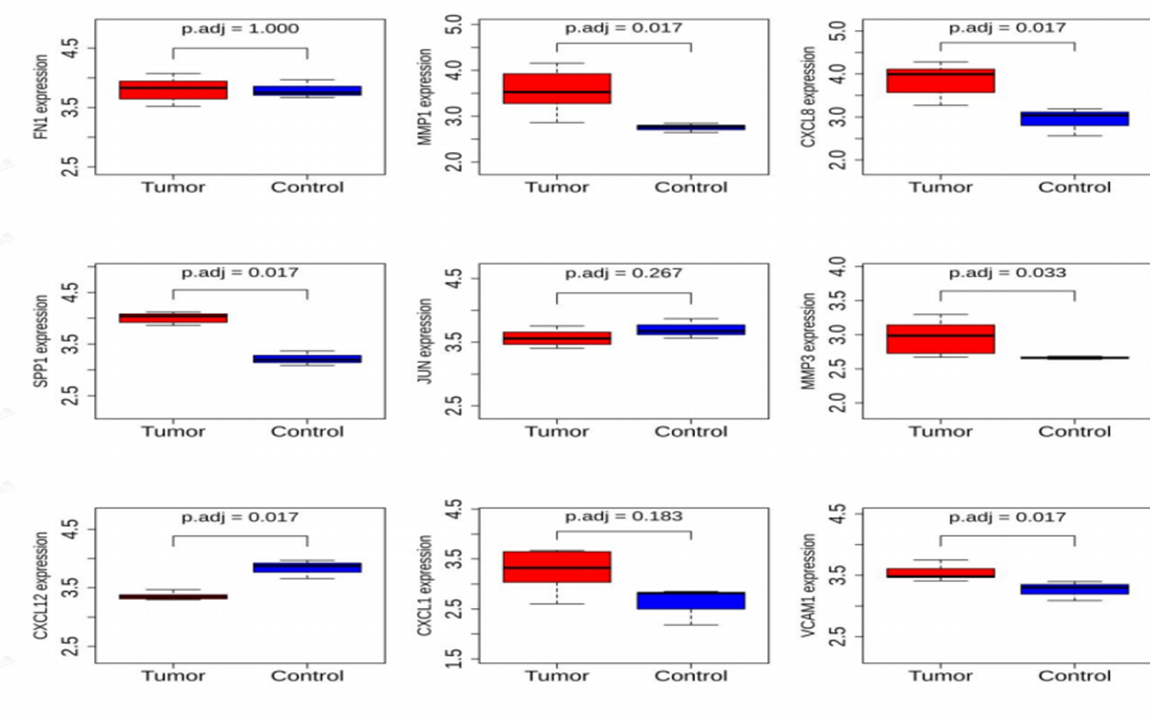
Figure 11: Expression analysis of Hub genes in external validation sets.
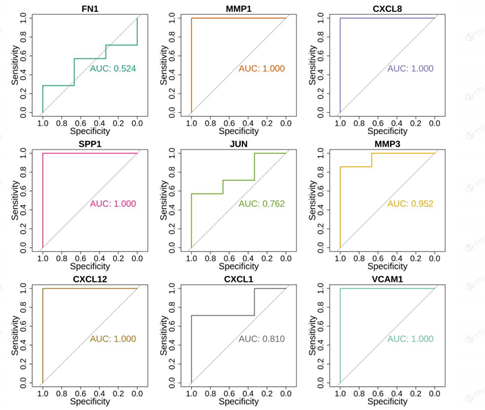
Figure 12: ROC analysis of Hub genes in external validation sets.
3.6 Hub Gene Survival Analysis
KM plots were drawn to illustrate the relationship between overall survival rate (OS) and gene expression levels in patients (Figure 13). Genes with p < 0.05 were defined as prognostic genes associated with survival by using the log-rank test. the results showed that five genes, MMP1, CXCL8, SPP1, MMP3, and VCAM1, were significant or defined as prognostic genes associated with survival. high expression of MMP1, CXCL8, SPP1, and MMP3 was associated with poor patient prognosis.
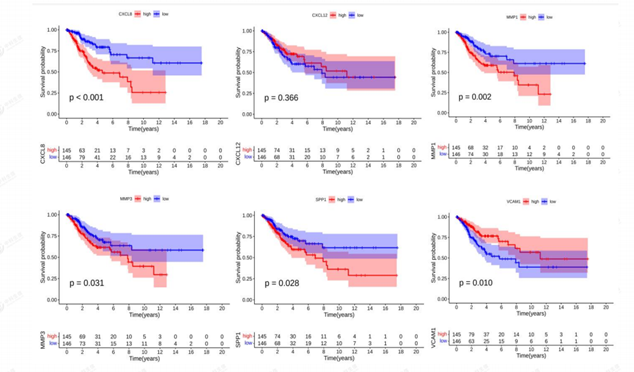
Figure 13: Kaplan-Meier of Diagnostic genes.
3.7 Correlation of diagnostic genes with immune infiltrating cells
A lollipop plot of each hub gene correlation with immune infiltrating cells was drawn using the R package ggpubr (Figure 14). The analysis of the
correlation between hub genes and immune infiltrating cells shows that CXCL8, MMP1, and SPP1 genes are all associated with immune cells such as CD4 T cells, macrophage M0, and macrophage M1.
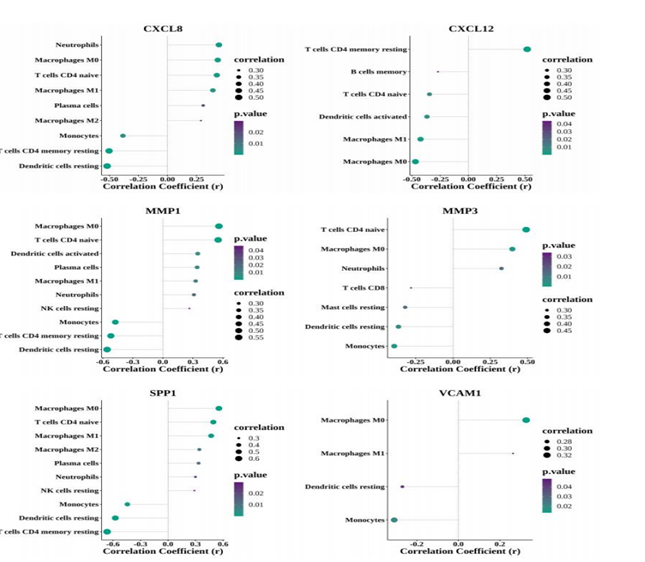
Figure 14. Correlation of diagnostic genes with immune infiltrating cells.
3.8 Regulatory mechanisms of diagnostic genes
Based on the results of diagnostic gene identification and validation, we mapped the Gene - TF and Gene - miRNA gene networks for diagnostic genes with their target miRNAs and TF transcription factors (Figure 15 and Figure 16. From the figure, it can be seen that CXCL8 has more transcription factors and also some of the same transcription factors among the 6 genes.
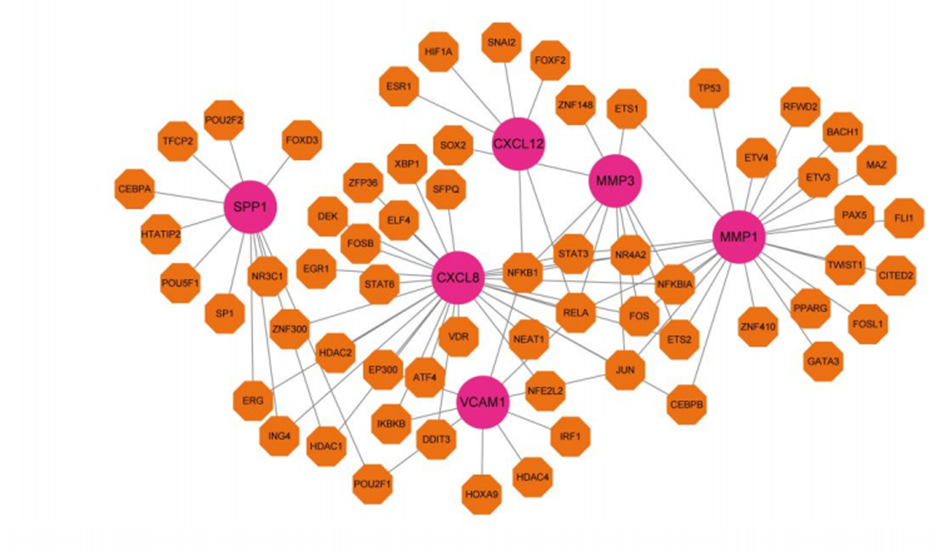
Figure 15: Gen-TF Gene Regulatory Network.
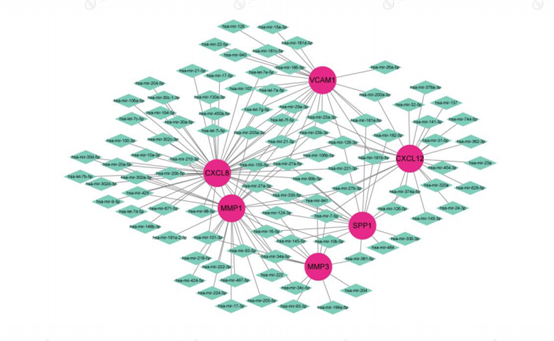
Figure 16: Gene-miRNA Gene Regulatory Network. Red: Diagnostic genes: Orange: TF: Green: miRNA.
There are many studies on genes associated with cervical cancer prognosis, but few studies have examined the genes associated with EMT in cervical cancer patients. For this reason, it is crucial to search for differentially expressed genes associated with EMT in cervical cancer. In order to find targets related to EMT regulation in cervical cancer to improve cancer invasion and metastasis, we screened the difference genes between cervical cancer and normal tissues based on the GSE9750 dataset.
We intersected 1440 differentially expressed genes with EMT-related genes, obtained 46 EMT-related differential genes, and performed GO and KEGG enrichment analysis on 46 genes. Reviewing the literature16, IL-17A and HPSE may promote cervical cancer tumor angiogenesis and cell proliferation and invasion through the NF-kB signaling pathway. KEGG enrichment pathway results show that CXCL8, CXCL12, VCAM1 are enriched in the NF-KB signaling pathway, and are all key genes in the progression of
cervical cancer EMT, this study has confirmed that CXCL8 and CXCL12 are highly expressed in cervical cancer patients, and their high expression is closely related to the poor prognosis of patients. Therefore, these EMT-associated differential genes may regulate cervical cancer by promoting cell proliferation and invasion.
To further search for key genes that influence cervical cancer, we screened the core module by PPI, ROC to evaluate the performance of diagnostic models. Subsequently, TF and miRNA networks related to diagnostic genes were constructed. Matrix metalloproteinase 1 (MMP1), which degrades the extracellular matrix, causes invasive metastasis and infiltration of malignant tumors17.Studies have shown18. that MMP1 plays a key role in regulating cervical tumor growth and lymgnostic factor in cervical cancer. Consistent with the results of this study by raw letter analysis. With increased MMP1 expression in patients, survival analysis revealed a decrease in OS, indicating that this gene may serve as a landmark for determining prognosis and be an excellent predictor of 5-year survival in CESC. It was shown19 that MMP1 may control macrophage activation of STAT3 pathway to promote tumor progression. MMP1 is more associated and positively correlated with macrophage M0 and T cells CD4 naive, and negatively correlated with the dendritic cells, CD4 memory T cells, and monocytes. Lower risk scores have been demonstrated20-23to be associated with larger levels of CD8+ T cells and activated CD4+ T cells, both of which are well known to be effector cells in the tumour microenvironment and typically have a better prognosis for patients. Therefore, these MMP1s may regulate the initiation and progression of cervical cancer by participating in the tumor immune microenvironment. Matrix metalloproteinase 3 (MMP3) plays multiple roles in extracellular protein hydrolysis and intracellular transcription. Knockdown of MMP3 has significant anti-tumor effects, such as inhibition of tumor cell migration and invasion. High levels of MMP3 expression are associated with poor prognosis in specific types of cancer (including head and neck, lung, pancreatic, cervical, gastric and uroepithelial cancers)24.This study also verified that the high expression of MMP3 in cervical cancer patients is associated with poor prognosis. Chemokine (CXCL8), also known as interleukin-8 (IL-8). CXCL8 expression in various tissues from the UCLCAN database showed that CXCL8 was upregulated in a variety of tumor types, including colorectal, cervical, esophageal, and head and neck cancers25, which is consistent with the results of this study regarding CXCL8.Secreted phosphoprotein 1 (SPP1), which controls cell growth, proliferation, migration and apoptosis. It has been shown26 that SPP1 expression is positively correlated with the abundance of M2 macrophages and the expression of the corresponding immune markers (CD163 and VSIG4).M2 macrophages exert anti-inflammatory and pro-tumor effects, promoting the progression and metastasis of a variety of tumors, such as breast and gastric cancers. Song et al27 noted that SPP1 upregulation was associated with cervical cancer invasion. This study confirmed the involvement of SPP1 in the progression of EMT in cervical cancer patients, which was positively correlated with macrophage M2, CD4 T cells, etc. In addition, its high expression was significantly associated with poor prognosis in cervical cancer patients. Vascular cell adhesion molecular 1 (VCAM1), an important member of the immunoglobulin superfamily, contains seven extracellular Ig structural domains that are normally detected in endothelial cells and activated by inflammatory factors28.A related study confirmed that VCAM1 is associated with the development of malignant tumors such as breast cancer, melanoma, and renal clear cell carcinoma .One study confirmed29 that high expression of VCAM1 enhanced the migration and invasive ability of cancer cells in vitro and affected the survival prognosis of patients, contrary to our raw letter findings. It was demonstrated30 that a variety of immune cells possess VLA-4, the particular receptor for VCAM1. Theoretically, tumor cells that express VCAM1 facilitate the ability for immune cells to connect to them and activate the immune system. The role of VCAM1 in tumor immunity deserves to be studied, which is why we subsequently performed the correlation between diagnostic genes and immune infiltrating cells. However, tumor patients have progressively lower immune function as their disease progresses, and their immune function transforms from cellular to humoral immunity while their mucus factors, such as VCAM1, are highly expressed. It has been shown31 that VCAM1 promotes colorectal cancer metastasis to lung or bone metastasis by recruiting monocytes or macrophages and forming complexes that promote tumor cell evasion from immune system attack and transendothelial cell migration .It may be deduced from the findings of this study that VCAM1 can facilitate the invasion and migration of cervical cancer cells by promoting tumor cell evasion of the immune system and transendothelial cell migration. Based on the information provided in this study, we look forward to further validating the specific mechanism of VCAM1's involvement in the EMT process.
There are a few issues with this study. There were no relevant animal trials or independent cervical cancer samples to validate the function of these five diagnostic genes. The impact of high VCAM1 expression on patient prognosis was also unforeseen and will be further investigated in future research. In addition, this study preliminarily confirmed that the six EMT-related characteristic genes screened are associated with tumor immune cells, but the specific mechanism of action of these diagnostic genes and the tumor immune microenvironment has not been confirmed. In the future, we will further examine whether these 6 genes may regulate immune cells.
In summary, this study identified 6 characteristic genes associated with EMT and identified many key genes associated with cervical cancer. Among them, 5 genes (MMP1, CXCL8, SPP1, MMP3, VCAM1) are closely related to the occurrence of EMT in cervical cancer patients, which can make a clearer diagnosis for clinically clear localization of cervical cancer patients' disease progression. Abnormal expression of these characteristic genes seriously affects the survival time of patients, and also confirms that these 5 genes are closely related to the immune response and the microenvironment of tumor immunity, suggesting that we may be able to improve the prognosis of cervical cancer patients through targeted immunotherapy. On the other hand, our next step will be to use TF and miRNA networks associated with diagnostic genes to explore the specific mechanisms of these genes in cervical cancer.
Supported by Guizhou Provincial Science and Technology Projects (2021) 450
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
