
Case Report | DOI: https://doi.org/10.31579/2641-0419/169
*Corresponding Author: K.V Charan Reddy, Department of Clinical and Interventional cardiology, Lilavati Hospital and Research Center, Mumbai, India.
Citation: Ajit Menon., Parag Bhalgat., K.V Charan Reddy., Abhishek Shah., Sanjeev Vichare., (2021) Approach to Asymptomatic Case of Bicuspid Aortic Valve with Coarctation and Massive Aortic Arteriopathy: a Ticking Time Bomb. J. Clinical Cardiology and Cardiovascular Interventions, 4(10); Doi:10.31579/2641-0419/169
Copyright: © 2021 K.V Charan Reddy, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 17 April 2021 | Accepted: 13 May 2021 | Published: 19 May 2021
Keywords: aortic coarctation (coa); bicuspid aortic valve (bav); arteriopathy; endovascular therapy
The combination of aortic coarctation (CoA) and bicuspid aortic valve (BAV) is associated with high risk of ascending aortic dilatation and type A aortic dissection. Most of the patients having combined lesions remain asymptomatic until adulthood and are incidentally diagnosed. Several questions regarding their treatment strategies remain unanswered. Both single-and two-stage surgical procedures have been described in medical literature, to treat CoA and BAV. Lately, combined endovascular therapy (Balloon angioplasty or Stent implantation) and surgery, has become a good alternative in selected cases. Here, we describe such a asymptomatic case of BAV with massive aneurysmal root dilatation and CoA, which was managed with endovascular stenting and surgery.
Bicuspid aortic valve (BAV) is the most common congenital heart abnormality, with a prevalence of 1-2%, and 3:1 male predominance [1]. It may present as an isolated cardiac defect or in association with other congenital heart malformations, such as coarctation of the aorta (CoA), aortic aneurysm or aortic dissection. Isolated form of CoA often occur as discrete stenosis or as hypoplastic segments of the ascending aorta, and account for 5-8% of total congenital heart disease (CHD) with prevalence of 3 in 10,000 live births [2]. Both BAV and CoA as congenital heart disease (CHD) are commonly associated in 85% of cases. Even though BAV is the most common CHD, only about 7% of patients with BAV have a concomitant CoA. On the contrary, BAV can be found in 70-75% of patients with CoA [3].
Though, both surgery or endovascular therapies have been described, vascular reactivity and mechanical properties of large conduit arteries may still remain impaired despite successful repair, due to early “programming” of vascular reactivity in utero. Furthermore, the management is based on anatomical considerations and complexity of lesion. Here, we describe the clinical profile and problems encountered in the management of a young asymptomatic adult with BAV, CoA and massive aortic root dilatation.
A 37 year-old male was referred for evaluation of hypertension. He gives a history of hypertension since the last 5 years and on regular medication of Amlodipine (5 mg/day). On examination, bilateral radial pulse was regular with very brisk upstroke, large amplitude, and rapid collapse. Bilateral lower extremity pulses were non-palpable. Blood pressure in both arms was equal at 160/40 mmHg. On auscultation of chest, S1 was normal and S2 was soft with a decrescendo, early-diastolic blowing murmur, best heard on the left lower sternal border (3rd and 4th intercostal spaces). The intensity of murmur increased on sustained hand grip. There was also suprasternal thrill and a continuous vascular murmur heard over the back.
Electrocardiogram (ECG) showed left ventricular hypertrophy. Chest X-ray showed mediastinal widening with cardiomegaly. Transthoracic Echocardiography (TTE), showed thickened, calcified biscuspid aortic valve (RCC-LCC fusion) with moderate to severe aortic regurgitation. AR pressure half time (PHT) was 195ms with regurgitant fraction (RF) of 48 mL. Left ventricular ejection fraction (LVEF) was 50% with no regional wall motion abnormality. Mitral valve was normal. Pulmonary hypertension was mild (38 mmHg). There was aneurysmal dilatation of the aortic root (36 mm), intersinus (91 mm) and proximal ascending aorta (82 mm) with narrowing just distal to left subclavian artery and gradient of 8 mmHg with diastolic run-off. CT-angiography confirmed 2D-ECHO findings (Figure 1). There were no intracranial aneurysms. Coronary arteries were normal

In view of the severity and complexity of the lesions, and to reduce the surgical risk and on-pump surgery time, combined endovascular therapy (Balloon angioplasty or Stent implantation) for CoA and subsequent surgery involving aortic valve with root replacement was planned.
Procedures
(a). Endovascular procedure
Under local anesthesia, 6Fr right femoral and 5Fr left radial access were secured. Baseline aortic arch pressure was noted as 140/80 mmHg and descending aorta beyond the CoA segment was 80/50 mmHg. The coarctation segment could not be crossed retrogradely even after repeated attempts. The coarctation segment was then crossed with a glide wire over 5Fr JR 4.0 diagnostic catheter antegradely from the left radial artery, and snared and externalized through the right femoral artery. A multipurpose catheter was then advanced over the wire, and placed in left subclavian artery. Catheter was then manipulated, and advanced into right subclavian artery and with the help of Amplatzer super stiff wire. Coarctation segment was predilated with a 6 x 20 mm balloon at 4 atm and stented with 8x45 mm CP stent (Figure 2).
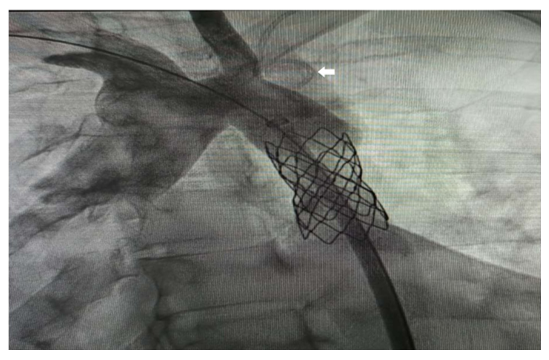
Haemodynamics improved immediately and pressures across the aortic arch and descending aorta was now 130/80 mmHg and 126/76 mmHg respectively. The patient was posted for surgery 3 days later.
(2).Surgical procedure
After sternotomy and routine cardiopulmonary bypass, the chest cavity was opened, revealing a large aorta with root diameter of about 9.2 cm (Figure 3a). The aortic root was dissected to visualize a incompetent and severely calcified biscuspid aortic valve (Figure 3b). There was also a healed aortic dissection at the aortic root. The aneurysmal segments including the root, proximal part of the ascending aorta along with aortic valve were excised. A 23 mm sized St Jude mechanical valved conduit was sutured in place. Individual coronary buttons (RCA and LCA) were attached to this conduit (Figure 3c). Patient made an uneventful recovery and was discharged seven days after surgery.

The combined pathologies of aortic CoA and BAV are associated with a higher risk of ascending aortic dilatation and type-A dissection. The presence of a bicuspid valve increases the risk of dissection by least ninefold, compared to a tricuspid aortic valve4. In the presurgical era, dissection of the aorta caused death in 19% of cases with coarctation, increased to 50% when coarctation coexisted with bicuspid aortic valve. The rate of expansion of aorta may vary considerably in BAV patients, but a diameter increase of 1-2 mm/year has been most commonly reported. However, a rapid progression up to 5 mm/year may occur and is associated with an increased risk of aortic dissection. The change in aortic diameters is influenced by the severity of valvular lesion (aortic stenosis or regurgitation) present in most BAV patients.
Nonetheless, quite a few young patients may experience a fast increase in aortic diameters despite echocardiographic normal BAV [5]. Patients with echocardiographic normal valves and diagnosis of BAV will require cardiovascular surgery in about 27% of the cases over a period of 20 years of follow-up [6]. The predominant hemodynamic lesion in BAV is related to the age of presentation, cuspal fusion patterns, and flow dynamics. The distribution of the different BAV fusion types are also important to study the form of resulting aortic dilatation. In LCC-RCC fusion type of BAV the ascending aorta is dilated along with the aortic root. In RCC-NCC fusion, dilatation of the distal ascending aorta is more common, and the involvement of the root is less frequent. Most of the patients having CoA remain asymptomatic until adulthood and are incidentally diagnosed during a investigation for systemic hypertension. 10% of CoA cases are diagnosed after 40-years of age and other cardiac pathologies accompany in ~40% of these cases. The cause of mortality in 20% cases of undiagonised CoA is sudden aortic rupture [7].
Current guidelines define the presence of aortic aneurysm at aortic diameter ≥40 mm or indexed aortic diameter ≥27.5 mm/m² in subjects with a short stature. Recommendations to operate isolated aortic dilation (i.e., without concomitant valvular lesion), is 55 mm in patients with BAV without additional risk factors such as arterial hypertension, family history of aortic dissection or progression of dilation above 3 mm/year. The cut-off value in patients with above mentioned risk factors is 50 mm, while 45 mm cut-off is common for patients if concomitant aortic valve surgery is scheduled [8].
Management of BAV and CoA has undergone major advances during the last decade. Traditionally, surgical treatment of a BAV and CoA was often performed in two steps. First step involved treating the critical CoA and collateral arteries through a left thoracotomy and second step involved the management of BAV and aortic dilatation with a medial sternotomy. It was a preferred method because of the high hemodynamic load and cardiac risk associated with both these conditions.
The choice of surgical techniques for CoA depends on evaluation of the lesion and surgeon’s experience. Resection of the CoA segment and end-to-end anastomosis was the most favoured method. Lately, a new single-stage procedure has been reported to be equally safe [9]. In this operation, after medial sternotomy, a extra-anatomic bypass from the ascending aorta to the descending aorta is created and BAV and COA repaired. However, it involves extensive dissection and increases length and complexity of procedure. Mobilization of descending aorta and end to side anastomosis with proximal arch can be done in neonates, but not in adults.
With the advent of endovascular therapy (Balloon angioplasty or Stent implantation), for the treatment of CoA, has lead to reduction in surgical morbidity and mortality. However, complex kinking of the aorta and histological changes in the aortic wall restricts its usage. Currently, endovascular therapy is the treatment of choice in children or older patients with multiple comorbidities [10].
The BAV and CoA are common diseases in the spectrum of CHD and are closely related to aortopathy, which must be included in all decision-making processes. Dilatation of the root and ascending aorta carry a significant risk of dissection in both diseases, but is highest in the combination of both. Aortic diameter is the most important parameter on which the indication for preventive aortic surgery is based. Rapid aortic growth (>3-5mm/year) is a risk factor for future ascending aortic events and is an indication to consider early surgery. Regular follow-up is advised and aortic lesions treated upon reaching certain cut-offs, before they become symptomatic. Based on anatomical complexities and surgeon’s experience, single or two-stage operation or combination of endovascular approach and surgery, have been described to safely treat concomitant BAV and CoA.
The authors declare no conflict of interest.
The authors are thankful to the patient for consenting to publish this case report.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
