
Research Article | DOI: https://doi.org/10.31579/2692-9759/133
Department of Cardiology, The Fourth Affiliated Hospital of Soochow University, Suzhou City, Jiangsu Province, P. R. China.
*Corresponding Author: Xiaofei Mei, MD, Department of Cardiology, The Fourth Affiliated Hospital of Soochow University, Suzhou City, Jiangsu Province, P. R. China.
Citation: Ziyin Huang, Yufeng Jiang, Yafeng Zhou, Xiaofei Mei, (2024), Analysis of Gut Microbiome Dysbiosis in Acute Myocardial Infarction Patients, Cardiology Research and Reports, 6(4); DOI:10.31579/2692-9759/133
Copyright: © 2024, Xiaofei Mei. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 29 July 2024 | Accepted: 06 August 2024 | Published: 13 August 2024
Keywords: acute myocardial infarction; atrial fibrillation; gut microbiota; correlation analysis
Background and Aim: This study aims to detect the structure of gut microbiota in acute myocardial infarction (AMI) patients complicated with newly diagnosed atrial fibrillation (AF) through high-throughput sequencing and LC–MS Based Metabolomics so as to explore the characteristic changes of gut microbiota in these patients.
Methods and Results: This prospective research recruited AMI patients, in the Fourth Affiliated Hospital of Soochow University. Patients who underwent physical examination were also included in this study. The characteristic of gut microbiota was conducted by high-throughput sequencing and LC–MS Based Metabolomics. At genus classification level, the abundance of Prevotella (P=0.009), Parabacteroides (P=0.036) in AMI patients with newly diagnosed AF were lower than that in AMI patients, while the abundance of Bifidobacterium (P=0.018), Dorea (P=0.036) Eggerthella (P=0.013), Peptoniphilus (P=0.039), Acinetobacter (P=0.016) tended to be higher. The species diversity of AMI patients with newly diagnosed AF was lower than that of AMI patients (P=0.037). Beta diversity analysis revealed a highly significant separation. The results of correlation analysis between differential metabolites and differential microbiota showed that the decrease of Prevotella and Parabacteroides in AMI patients with newly diagnosed AF may lead to an increase in some metabolites such as N-Acetyltyrosine, L-Isoleucine, 7-Methylguanine, L-Tyrosine, Sphingosine, 6-methoxyquinoline and 4-Hydroxybenzaldehyde. These changes will influence multiple metabolic pathways, thus affecting the prognosis of patients.
Conclusions: There was an imbalance of gut microbiota in AMI patients, including AMI patients with newly diagnosed AF.
Acute myocardial infarction (AMI) is the main cause of cardiovascular death which entails serious concerns [1-2]. The most frequent arrhythmia in AMI patients is atrial fibrillation (AF). AMI patients with newly diagnosed AF are at higher risk of reinfarction, strokes, heart failure, and sudden cardiac death [3]. Therefore, the treatment of AMI patients with newly diagnosed AF has always been a hot spot in the field of cardiovascular research [4-6].
The gut microbiota resides in hosts’ intestine and participates in various pathological and physiological processes of hosts. Current research shows that the dysbiosis of gut microbiota has been proposed to be a pathogenic factor in several diseases such as diabetes, hypertension, atherosclerosis and thrombotic event [7-10]. So far, many articles with regard to cardiovascular disease and gut microbiota have been published [11-13]. While the association between gut microbiome and AMI with newly diagnosed AF is still lack of relevant research. This study aims to detect the change of gut microbiota in AMI patients with newly diagnosed AF through high-throughput sequencing and metabonomic analysis. We suggest the findings from this study will be clinically beneficial for the diagnosis and treatment of AMI patients with newly diagnosed AF.
Study population
From August 2021, the research has recruited three AMI patients with newly diagnosed AF (group A), nine AMI patients (group B) and eighteen patients (group C) who underwent physical examination in the same hospital. The study excluded patients who take antibiotics or ingest probiotics within three months; patients using antibiotics in skin within one month; and patients with intestinal diseases or a history of bowel surgery [14].
Sample collection and sample detection
Research subjects emptied their stool into special stool collection containers. After the defecation was completed, research subjects need to scoop up 5g deep stool and place it in the professional stool DNA preservation solution. The stool samples were stored at -80°C until further processing.
The DNA of gut microbiota were extracted by using the QIAamp DNA stool kit. We designed the target region and fusion primers according to the requirements of the sequencing platform, and used a two-step method for PCR amplification. The PCR amplification product obtained in the previous step were recovered by using 2% agarose gel. We used the AxyPrep DNA Gel Extraction Kit to purify the amplified PCR product. we used the Illumina sequencing platform to perform high-throughput sequencing on samples we collected. Finally, untargeted LC-MS based metabolomics research was carried out by MS2-NEG and MS2-POS analysis [15-16].
Data analysis
The composition of gut microbiota among three groups were presented at the classification level of phylum and genus. Wilcoxon rank sum test was used to compare the abundance of each microbiota when value distribution deviated from normality. When testing the differences of alpha diversity, T-test was used to compare three indexes between two groups. NMDS plots generated from weighted uniface distances were used to display the discrepancy between different groups. Untargeted LC-MS based metabolomics research was carried out by MS2-NEG and MS2-POS analysis. Finally, we conducted a correlation analysis between metabolites and species.
3.1 Composition of gut microbiota
This part mainly analyzed the gut microbiota composition of three groups at phylum and genus classification levels. As shown in the figure1, Firmicutes, Bacteroidetes and Proteobacteria dominated the gut microbiota composition of three groups at phylum level. At genus classification level, Bacteroides and Prevotella were two kinds of microbiota with the highest abundance. The most differentially abundant microbiota among three groups was identified by LEfSe analysis (figure 2).
At phylum classification level, the abundance of each species was different among three groups. The microbiota with statistically significant differences between AMI and AMI with newly diagnosed AF groups were Firmicutes (P=0.036) and Actinobacteria (P=0.018). While at genus classification level, the abundance of Prevotella (P=0.009), Parabacteroides (P=0.036) in AMI with newly diagnosed AF group were lower than that in AMI group, while the abundance of Bifidobacterium (P=0.018), Dorea (P=0.036) Eggerthella (P=0.013), Peptoniphilus (P=0.039), Acinetobacter (P=0.016) were higher.
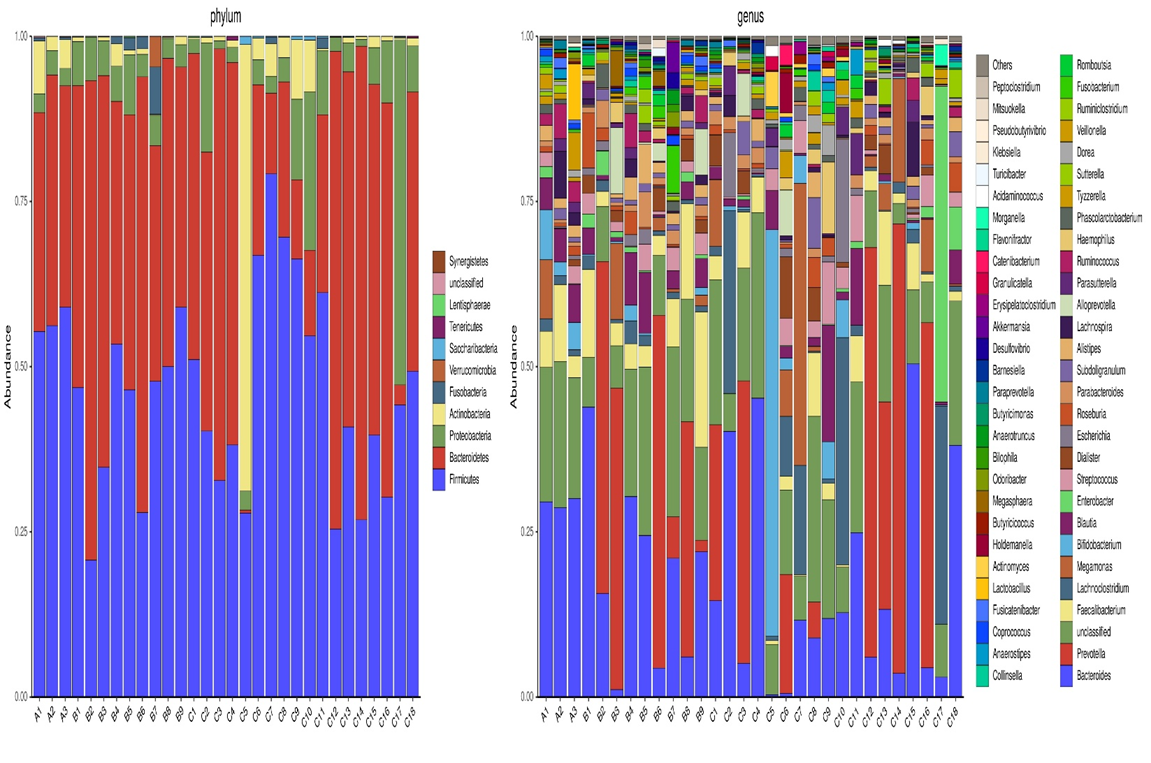
Figure 1: Differences of gut microbiota composition among three groups
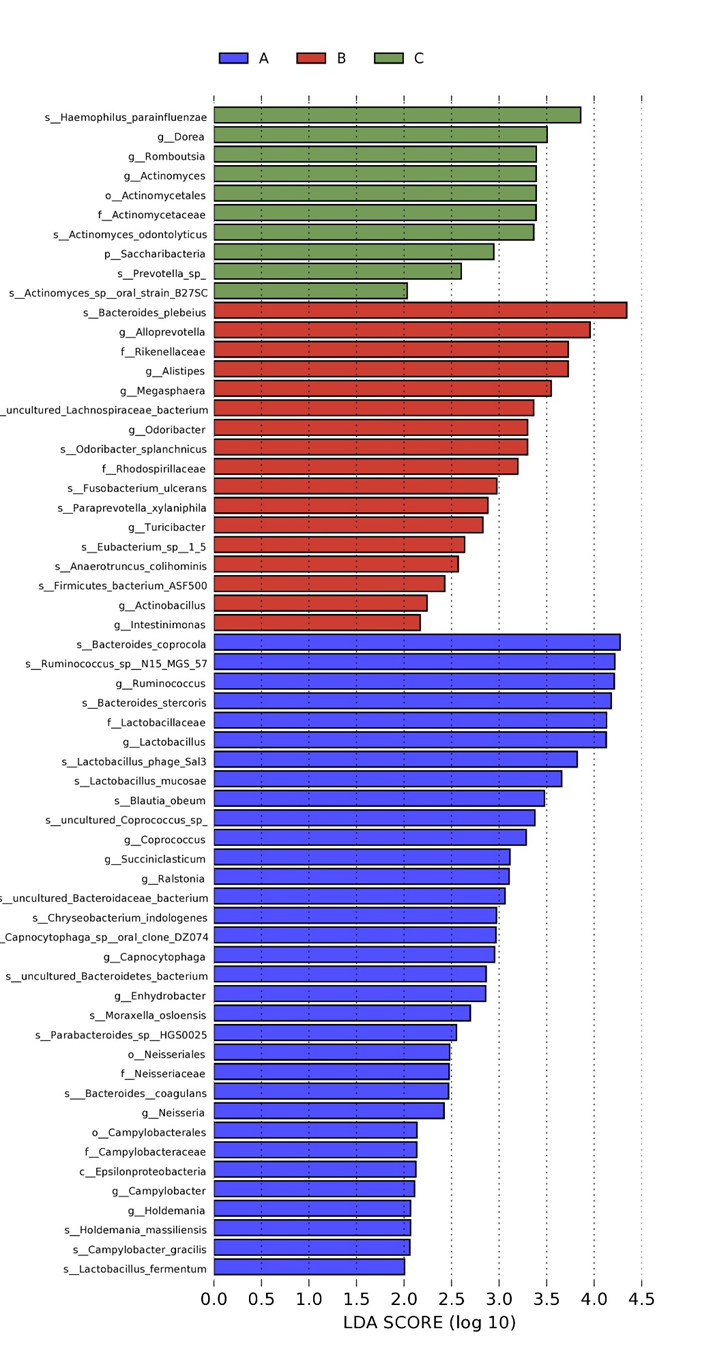
Figure 2: The results of LEfSe analysis.
3.2 The differences of gut microbiota with regard to alpha and beta diversity
With regard to alpha diversity, reflecting intra-individual variance, three indexes (ace index, chao index, simpson index) were evaluated. There was a difference in simpson index, indicating that the species diversity of three groups was different. Meanwhile, the species diversity of AMI with newly diagnosed AF group was lower than that of AMI group (P=0.037).
For beta diversity, NMDS plots generated from weighted unifrac distances were used to display the discrepancy between different groups (figure 3). Beta diversity analysis revealed a highly significant separation.
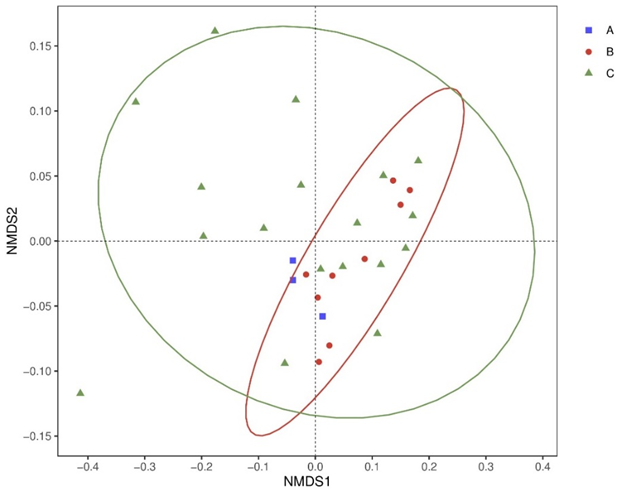
Figure 3: Beta diversity differed significantly among three groups
3.3 Untargeted LC-MS based metabolomics approach
In untargeted LC-MS based metabolomics approach, metabolomics with VIP (variable importance in projections) value greater than one can be regarded as differential metabolites. There were 20 metabolites with statistical differences between AMI and AMI with newly diagnosed AF groups based on MS2-NEG analysis. While according to the MS2-POS analysis, Hydroquinone, 4-Hydroxybenzaldehyde, Phenethylamine, 2-Phenylacetamide, L-Glutamic acid, gamma-Glutamylleucine, 1-AMINOCYCLOPROPANECARBOXYLATE, Oxypurinol, 6-methoxyquinoline, Chenodeoxycholyllysine, Sphingosine, NAE 20:1 and N-METHYL-D-ASPARTIC ACID were significantly different between two groups (figure 4).
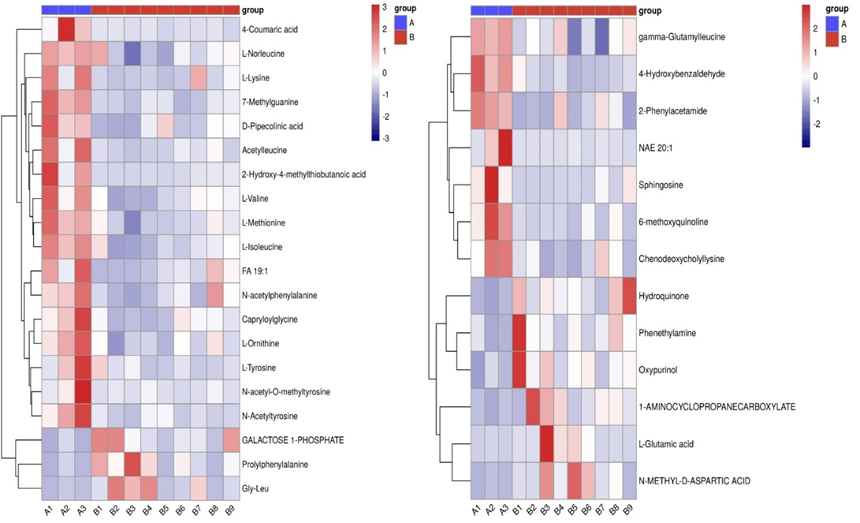
Figure 4: hierarchical clustering heat map between AMI and AMI with newly diagnosed AF groups.
3.4 Correlation analysis
Correlation analysis between differential metabolites and differential microbiota (genus level) were presented in the form of heat map (figure 5). Thermogram can reflect the correlation between microbiota and metabolites through color gradient. As shown in the following figure, the abundance of Prevotella was negatively correlated with 7-Methylguanine (P=0.002), N-Acetyltyrosine (P<0 P=0.002) P=0.005) P=0.004). P=0.007) P=0.004).>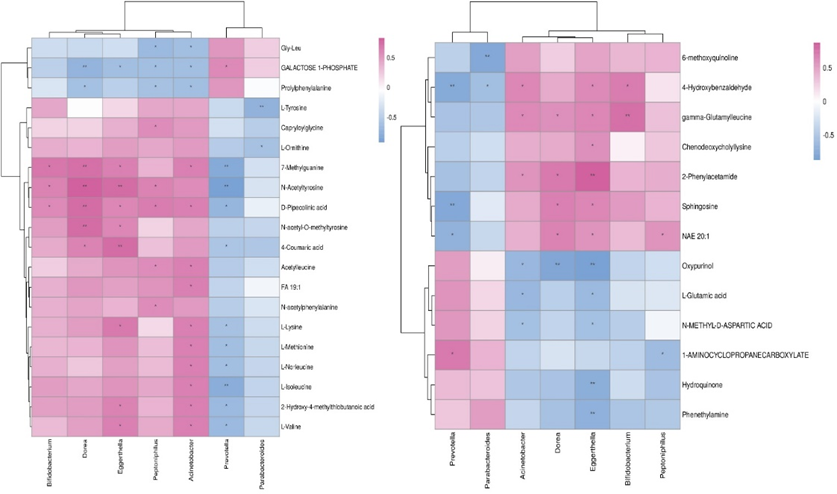
Figure 5: Correlation analysis between differential metabolites and differential microbiota between AMI group and AMI with newly diagnosed AF group.
The microbial community in the human gut includes trillions of interacting microbes, which plays an important role in affecting human health and diseases [17-19]. A theory called gut hypothesis of coronary heart disease (CHD) has attracted the attention of cardiovascular physician. While the association between gut microbiome and AMI patients with newly diagnosed AF is still lack of relevant research. Therefore, we plan to conduct a large-scale study to explore the characteristic changes of gut microbiome in AMI patients with newly diagnosed AF. This article presented the preliminary results of this study.
Through the preliminary exploration of gut microbiota in AMI patients with and without newly diagnosed AF, we have obtained many valuable results. Dysbiosis of gut microbiota has been found both in AMI patients and AMI patients with newly diagnosed AF. The species diversity of gut microbiota decreased most significantly in AMI with newly diagnosed AF group. The reduction of species diversity will affect normal physiological processes in patients, thus making AMI fall into a vicious circle [20-22].
In this study, we identified the differences of gut microbiota between the AMI patients, AMI patients with newly diagnosed AF and controls. At genus classification level, the abundance of Prevotella and Parabacteroides in AMI with newly diagnosed AF group were lower than that in AMI group, while the abundance of Bifidobacterium, Dorea, Eggerthella, Peptoniphilus, Acinetobacter were higher [23-25]. Parabacteroides is one of the main components of human gut microbiome. This microbiota has been proved to regulate mucosal immune system, reduce inflammation and participate in carbon metabolism. In addition, Parabacteroides can secrete short chain fatty acids such as acetate and propionate to affect the health of the host [26-28]. Together, the changes of gut microbiota in AMI patients with newly diagnosed AF was a decrease in the abundance of Parabacteroides which is associated with anti-inflammatory effects. This prolonged inflammatory state may have a bad impact on the prognosis of these patients. We further conducted a correlation analysis between differential metabolites and differential microbiota. We have shown that Parabacteroides was negatively correlated with L-Tyrosine and 6-methoxyquinoline. 6-methoxyquinoline is an intermediate product of glycolysis. In another words, the decrease of Parabacteroides in AMI patients with newly diagnosed AF will further activate the glycolysis pathway. Enhanced glycolysis leads to excessive accumulation of hydrogen ions and lactic acid in cardiomyocytes. This change can cause endoplasmic reticulum stress and mitochondrial dysfunction which will further aggravate myocardial injury [29-31]. While L-Tyrosine is one of the key substances in the methionine metabolism pathway. It is the precursor of bioactive substances such as catecholamine neurotransmitters (such as dopamine, norepinephrine and epinephrine), thyroid hormone and melanin. The increase of L-tyrosine in AMI patients with newly diagnosed AF will cause the activation of substances such as epinephrine, then affecting the cardiac remodeling [32-34]. In conclusion, these changes will influence the glycolysis pathway and amino acid metabolism, thus affecting the prognosis of patients [35-37]. However, the increase of 7-Methylguanine, N-Acetyltyrosine, L-Isoleucine, 4-Hydroxybenzaldehyde and Sphingosine caused by Prevotella have not been confirmed to be associated with AMI [38-40].
As the preliminary research of a large-scale clinical study, our research also obtained many valuable results. This study provided relevant information on the characteristic changes of gut microbiota in AMI patients with newly diagnosed AF. However, due to the limited sample size the results of this study need to be further verification and confirmation. We suggest that further research will be clinically beneficial for the specific diagnosis and treatment of AMI patients with newly diagnosed AF.
There was an imbalance of gut microbiota in AMI patients, including AMI patients with newly diagnosed AF. The changes of gut microbiota in AMI patients with newly diagnosed AF was a decrease in the abundance of microbiota which is associated with anti-inflammatory effects. And the species diversity of AMI patients with newly diagnosed AF tended to be lower. The results of correlation analysis showed that these changes will influence multiple metabolic pathways, thus affecting the prognosis of patients.
This study has been approved by the Fourth Affiliated Hospital of Soochow University Ethic Board and registered in Chinese Clinical Trial Registry (ChiCTR2100050121). Research subjects were assessed for eligibility and written informed consent.
None
ZH, YJ, and YZ designed the study. ZH wrote the first draft of this report. YZ and YJ helped to write the final version. All authors read and met the criteria for authorship and agree with the results and conclusions of the report.
This work was supported by grants from National Natural Science Foundation of China (81873486), the Science and Technology Development Program of Jiangsu Province-Clinical Frontier Technology (BE2022754), Clinical Medicine Expert Team (Class A) of Jinji Lake Health Talents Program of Suzhou Industrial Park(SZYQTD202102), Suzhou Key Discipline for Medicine(SZXK202129), Demonstration of Scientific and Technological Innovation Project (SKY2021002), Suzhou Dedicated Project on Diagnosis and Treatment Technology of Major Diseases(LCZX202132), Research on Collaborative Innovation of medical engineering combination(SZM2021014), Research on Collaborative Innovation of medical engineering combination (SZM2022003), Suzhou Key Laboratory of Diagnosis and Treatment of Panvascular Diseases(SZS2023021). Research on Collaborative Innovation of Medical Engineering Combination (SZM2022019).
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
