
Case Report | DOI: https://doi.org/10.31579/2690-4861/068
Dr. Ion Cantacuzino Clinical Hospital, University of Medicine and Pharmacy „Carol Davila”, Bucharest, Romania.
*Corresponding Author: Marilena Stoian, Dr. Ion Cantacuzino Clinical Hospital, University of Medicine and Pharmacy , Carol Davila”, Bucharest, Romania.
Citation: Marilena Stoian. (2020) Acute Renal Failure and Thrombocytopenia To a 36-Year -Old Woman: Case Report. International Journal of Clinical Case Reports and Reviews. 4(3); DOI: 10.31579/2690-4861/068
Copyright: © 2020 Marilena Stoian, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 26 September 2020 | Accepted: 05 November 2020 | Published: 09 November 2020
Keywords: antiphospholipid syndrome; thrombophilia; antiphospholipid antibodies; renal biopsy; aps nephropathy
Background: Antiphospholipid syndrome (APS) is an acquired, immune-mediated thrombophilia occurring alone (primary APS, PAPS) or in association with other autoimmune diseases, mainly systemic lupus erythematous (SLE), (secondary APS), characterized by recurrent venous or arterial thrombosis and /or pregnancy morbidity in association with antiphospholipid antibodies (aPL) and/or lupus anticoagulant (LA).
Case report: A 36-year-old woman was admitted to the hospital because of acute renal failure and thrombocytopenia. This woman presented with an acute illness characterized by a prodrome of respiratory symptoms and fever that were unresponsive to antibiotic therapy, followed by the progressive involvement of multiple organs. There was enzymatic and functional evidence of myocardial necrosis leading to intermittent, severe heart failure, as well as acute renal failure requiring dialysis, laboratory evidence of pancreatic injury, pulmonary infiltrates with respiratory failure, and central nervous system involvement, with confusion. After a renal biopsy was detected a perinephric hematoma and thrombocytopenia. The laboratory criteria for the diagnosis of the antiphospholipid-antibody syndrome are an elevated value for IgG anticardiolipin antibody, a positive test for lupus anticoagulant, and an elevated value for IgM anticardiolipin antibody.
Conclusions: This patient has the antiphospholipid antibody syndrome, with an acute thrombotic angiopathy that caused ischemic damage in the myocardium, pancreas, kidneys and lungs. The renal interstitial inflammation is more severe than that expected from the ischemic injury alone and many reflect either a hypersensitivity drug reaction or in view of the dry Sjogren’s syndrome.
Antiphospholipid syndrome (APS) is a chronic, systemic, autoimmune disease characterized by recurrent venous or arterial thrombosis and /or pregnancy morbidity in association with antiphospholipid antibodies (aPL) and/or lupus anticoagulant (LA) [1]. Pregnancy morbidity is defined as one of more unexplained fetal deaths, of a morphologically normal fetus after the 10th week of gestational age, or three or more premature labours (before the 34th week of gestational age) due to pre-eclampsia, or eclampsia , or three or more abortions in the absence of anatomic, hormonal, or chromosomal anomaly of the parents.aPL are IgG or IgM antibodies detected by enzyme-linked immunosorbent assay(ELISA) using the negatively charged phospholipid cardiolipin as the antigen (anti-cardiolipin (aCL) antibodies).The majority of the aPL recognize a plasma apoprotein, known as β2 - glycoprotein 1 (β2GPI) either alone or in complex with phospholipids[2].ELISAs detecting anti β2GPI antibodies in a phospholipid free fashion have shown that anti- β2GPI antibodies are a rather specific marker for thromboembolic events. [3]. LA represents a group of which inhibit in vitro the conversion of prothrombin to thrombin resulting in prolongation of the partial thromboplastin time (PTT);the result is not corrected by adding normal plasma[4].Antibodies of the LA type recognize mainly prothrombin. The diagnosis of definite APS is made when the patient fulfils one clinical (thrombosis or pregnancy morbidity) and one laboratory (aPL or LA) criterion [1]. APS is an acquired, immune-mediated thrombophilia occurring alone (primary APS, PAPS) or in association with other autoimmune diseases, mainly systemic lupus erythematous (SLE), (secondary APS) [5].Very infrequently, APS can be a severe disease characterized by microthrombi in the small vessels of many organs, and multi-organ failure (catastrophic APS).
Thrombosis in Aps
As our understanding of these antibodies increased there was hope that pathogenic mechanisms would be revealed and much attention was focused on β2GPI [6][7][8]. This apolipoprotein is composed of 326 amino acids, has multiple disulphide bonds, and a high carbohydrate content (19%) resulting in an apparent molecular weight of 50 kDa. The molecule has five repeating globular (or Sushi) domains each of about 60 amino acids with highly conserved disulphide bonds. It has several effects on the hemostatic system. It inhibits the contact activation of the intrinsic pathway of coagulation, interferes with the assembly of the prothrombinase complex on platelet membranes and phospholipid vesicles and also inhibits the analogous tenase complex. It increases adenylate cyclase activity in platelet membranes and inhibits the release reaction of platelets during ADP-induced aggregation [9][10]. If β2GPI were to act as a natural anticoagulant then perhaps antibodies to it could predispose to thrombosis. That these antibodies might have a pathogenic role is supported by some animal work. Naive mice immunized with β2GPI developed aCL. This was later followed by prolongation of the PTT, thrombocytopenia, and when the mice were mated, by a high rate of fetal resorption in utero [11]. There is, however, no direct evidence that β2GPI acts as an anticoagulant and deficiency of β2GPI is not associated with thrombosis. Patients with antiphospholipid syndrome have auto-antibodies against proteins other than β2GPI and prothrombin. It is possible that the thrombotic manifestations are due to antibodies to proteins such as protein C, protein S, phospholipase A2, or annexin V, or to antibodies against endothelial cells. There is, however, no convincing evidence that any of these antibodies could explain the antiphospholipid syndrome. Galli and Bevers [12] have proposed that abnormal exposure of procoagulant phospholipid surfaces to the bloodstream may induce binding of plasma proteins with affinity for anionic phospholipids (such as β2GPI and prothrombin). Binding of these proteins could cause a conformational change and render them immunogenic, inducing the production of aPL. At the same time the pathological exposure of procoagulant surfaces promotes the phospholipid-dependent coagulation reactions and causes thrombosis. If this hypothesis is correct then aPL are just an epiphenomenon.
Case Report
A 48 year old woman was admitted to the hospital because of acute renal failure and thrombocytopenia. This woman presented with an acute illness characterized by a prodromal of respiratory symptoms and fever that were unresponsive to antibiotic therapy, followed by the progressive involvement of multiple organs. There was enzymatic and functional evidence of myocardial necrosis leading to intermittent, severe heart failure, as well as acute renal failure requiring dialysis, laboratory evidence of pancreatic injury, pulmonary infiltrates with respiratory failure, and central nervous system involvement, with confusion. A perinephric hematoma and thrombocytopenia were detected after renal biopsy.
Twenty years earlier had been diagnosed a prolonged activated partial thromboplastin time and she also had a history of lupus-like symptoms. During the current illness, a prolonged activated partial thromboplastin time was noted and positive test for antinuclear antibodies and also thrombopenia and anemia. [Table 1]
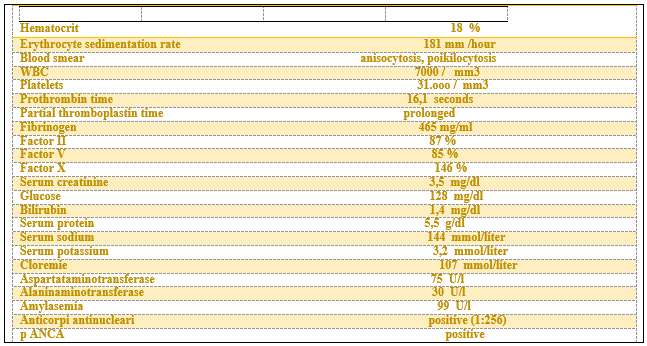
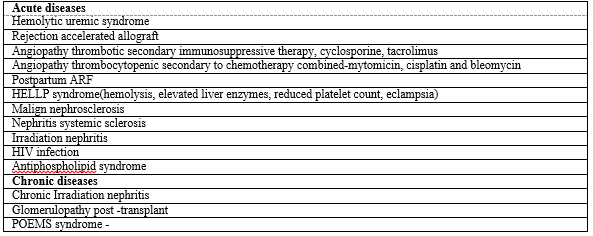
Examination of the renal biopsy specimen revealed abnormalities in all components of the cortex. The combination of hypercellularity of the glomerular capillary tuft and thickening of the capillary walls duplication defines the membranoproliferative pattern of glomerular injury. [Figure 1] Although the most frequent causes of this form of glomerular injury are diseases characterized by circulating immune complexes, which are either idiopathic or due to known autoimmune dis-eases or chronic infections, paraprotein deposition diseases and thrombotic angiopathies can result in virtually identical changes on light microscopy. The extensive injury of the arteries, arterioles and glomerular microvasculature suggests an acute exacerbation of a thrombotic angiopathy [Table 2]
Virtually all the typical changes of an acute thrombotic event are present in this patient kidneys endothelial swelling and degeneration, congestion and thromboses of glomerular capillaries, separation of the endothelium from the glomerular basement membrane and subendothelial accumulation of electron-lucent fluffy material, the absence of electron-dence deposits, and fibrinoid necrosis and thrombosis of arterioles and small arteries. The biopsy specimen also showed chronic vascular changes, including the typical duplication of the glomerular capillary wall, mesangial sclerosis, and proliferative changes in arteries and arterioles, resulting in an accumulation of subintimal mucoid connective tissue and the typical onionskin change.
The laboratory criteria for the diagnosis of the antiphospholipid-antibody syndrome are an elevated value for IgG anti cardiolipin antibody, a positive test for lupus anticoagulant, and an elevated value for IgM anti cardiolipin antibody. This patient has the antiphospholipid antibody syndrome, with an acute thrombotic angiopathy that caused ischemic damage in the myocardium, pancreas, kidneys and lungs. The renal interstitial inflammation is more severe than that expected from the ischemic injury alone and many reflect either a hypersensitivity drug reaction or in view of the dry Syogren syndrome.

(A) Well-demarcated, wedge-shaped area of sub-capsular tubular atrophy and thyroidization of tubules. This parenchymal change, known as focal cortical atrophy (FCA), is characteristically seen in APSN. (Hematoxylin and eosin, original magnification ×200.) (B) This shows typical pseudocystic or ballooned glomeruli in areas of FCA. One normal glomerulus is also seen in the surrounding normal parenchyma. (Hematoxylin and eosin, original magnification ×200.) (C) Typical vascular changes florid arteriolosclerosis with obliteration of the lumen of many arterioles in a case of APSN. (Hematoxylin and eosin, original magnification ×200.) (D) Thrombotic microangiopathy. Fibrin thrombi are present in arterioles at the hilum of the glomerulus and in many glomerular capillaries (Hematoxylin and eosin, original magnification ×400)
Neprhopathy of Aps (APSN)
In the context of thrombophilia renal involvement is expressed as tubulointerstitial or glomerular dysfunction, due to the occlusion of large, medium or small sized renal arteries [13]. However it is difficult to establish unique clinicopathologic characteristics of APS because:
renal involvement in primary APS occurs rarely;
renal involvement in secondary APS can be attributed to the underlying disease, mainly SLE;
The clinical characteristics of APS represent a continuum from PAPS to SLE.Therefore; renal disease may represent the first manifestation of SLE; the best strategy to study APS nephropathy related features is to evaluate isolated patients with PAPS and renal involvement [14].
Clinical features of APSN
Problems that have been associated with aPL are listed in Table3. Clinically the significant problems are thrombosis, fetal loss and to a lesser extent thrombocytopenia.

Hypertension
Hypertension is one of the first major features described in association with livedo reticularis and aPL [15]. It is exceedingly common in both primary and secondary APS, and is considered to be a sensitive marker of nephropathy in this syndrome. In the series of primary APS reported by Nochy et al., hypertension was present in 93% of their 16 patients and was sometimes the only clinical sign suggestive of nephropathy [16]. Consequently, it was recommended to investigate patients with APS complicated by hypertension for an underlying renal lesion. Imaging the renal arteries should be considered as RAS/thrombosis has been shown to respond well to anticoagulation with or without percutaneous balloon angioplasty, leading to recovery of renal function and return to normal blood pressure [17, 18]. Hypertension in APS may often be severe, with some patients presenting with hypertensive emergencies [19, 20]. Malignant hypertension in APS without overt lupus nephritis or stenosis/thrombosis of the main renal arteries was documented by Cacoub and colleagues in five patients. Interestingly, they used high-dose steroids and anticoagulation to treat the biopsy-proven microangiopathy, resulting in rapid resolution of the hypertensive crisis in three of their patients [19].
Renal artery lesions
APS has been well documented as a unique, non-traditional cause of RAS with important clinical consequences. It may manifest in multiple ways ranging from renal infarction to ischemic acute renal failure to slowly progressive ischemic chronic renal failure to renovascular disease. Using magnetic resonance angiography, 26% of aPL-positive patients with poorly controlled hypertension were found to have RAS, significantly higher compared with 8% of relatively young (≤50 years) hypertensive controls and 3% of healthy potential kidney donors [21]. Two patterns of stenotic lesions have been described in APS. The more common pattern is characterized by smooth, well-delineated and often non-critical stenosis in the mid-portion of the renal artery, quite distinct from either fibromuscular dysplasia or atherosclerosis. The less common pattern is similar to atherosclerotic lesions situated proximally and occasionally involving the aorta [21]. Although the nature of the RAS in APS remains unclear, the response to anticoagulation does suggest a thrombotic process. Previous reports have shown that anticoagulation treatment with international normalized ratio (INR) >3 may reverse RAS and achieve subsequent clinical improvement [22, 23]. Remondino et al. [24] reported a case of bilateral RAS associated with APS in which complete recanalization of both renal arteries was observed on a repeat renal spiral CT scan after 5 months of treatment with anticoagulation. Besides thrombosis, other potential etiologic factors for RAS in APS include accelerated atherosclerosis [25] as well as increased endothelin levels resulting in vasoconstriction [25, 26].
Renal infarction
Renal infarction results from occlusive lesions in smaller diameter intraparenchymal vessels, which are caused by either in situ thrombosis or emboli from a pre-existing upstream arterial lesion or a cardiac valvular lesion [17]. Clinically, patients with renal infarction, which could be the first manifestation of APS, may present with flank pain, severe hypertension and/or renal dysfunction. Some of these patients may have multiple, often serious, thrombotic episodes, and many have multiple infarctions in the renal cortex [27]. Histologically, glomerular ischemia, tubular atrophy and interstitial fibrosis may be seen in patients with renal infarction due to primary APS. Interestingly, in five out of eight such patients in whom biopsies were performed, no histological evidence of associated thrombotic microangiopathy (TMA) was found, as summarized in the report by Poux et al. [28]. Although renal infarction is a rare complication of APS, this diagnosis should be considered in any young subject who presents with renal infarction.
Histopathologic features of APSN
The most characteristic histopathologic lesion of PAPSN is a thrombotic microangiopathy of the kidney (31%) combined with rather chronic fibrotic vascular lesions as: arteriosclerosis (75%); fibrous intimal hyperplasia (75%); arteriolar occlusions (68%); focal cortical atrophy (62%) [29] However, the renal findings of APSN are not pathognomonic. Glomerular thrombosis occurs in SLE nephritis, in the absence of APS [30], while intense vascular inflammation has been observed in PAPS related renal involvement [31][32]. In addition, co-occurrence of APSN with SLE related nephropathy is a reason for deterioration of the renal function [33]. [Figure 1]
Pathogenesis of APSN
There is indirect evidence that aCL antibodies recognize B2 GPI on the endothelial cell membrane, resulting in transmembrane signaling with the following consequences:
Increased expression of adhesion molecules like intracellular adhesion molecule-1(ICAM-1), vascular adhesion molecule-1 (VCAM-1) and E-selectin;
Transient expression of tissue factor (TF) on endothelial cell and peripheral monocytes, leading to the initiation of the exogenous coagulation cascade;Occasionally the apoptotic death of endothelial cells [34].
Increased expression of adhesion molecules results in increased adhesion of monocytes on endothelial cell surface and this may explain the intense vascular inflammation found in the intrarenal arteries and the mesangium in case reports of APS nephropathy[31][32].
Therapy of APSN
To establish the appropriate therapy renal biopsy is mandatory. In case the nephritis can be classified according to the WHO classification system for lupus nephritis, the patient should receive the usual therapies for lupus nephitis.if the histologic findings indicate thrombi or fibrous intimal hyperplasia, either, with, or without proliferative changes, the patients should receive throughout life, anticoagulants in addition to cytotoxic therapy.
Conclusions
APS is being increasingly recognized as an important cause of renal injury due to thrombosis at any location within the renal vasculature. Accordingly, the renal manifestations of APS may include systemic hypertension in association with livedo reticularis, renal artery lesions, renal infarction, APSN, renal vein thrombosis and increased allograft vascular thrombosis. Testing for aPL must therefore be considered in patients with any of these manifestations. Nephrologists are expected to be involved more frequently in managing patients with APS, whether it is primary, secondary or, most certainly, with CAPS. Renal pathologists should carefully examine renal biopsies obtained from SLE patients with positive aPL for the presence of APSN, as this may have significant implications on therapeutic decisions. Biopsy proven renal involvement in PAPS accounts for one-tenth of patients The most characteristic histopathologic finding of APSN is fibrous intimal hyperplasia. When APS related nephritis complicates SLE nephritis, the course could be rather severe and the renal function could deteriorate Anticoagulation remains the mainstay treatment of patients with renal involvement due to APS. In addition, patients with catastrophic features often require immunosuppressive therapy. Future studies may help to identify more targeted therapeutic agents.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
