
Case Report | DOI: https://doi.org/10.31579/2578-8949/003
*Corresponding Author: Cesar Ricardo, Department of Dermatology, Sweden.
Citation: Cesar Ricardo, Brady Mark, Diego Luis, A unique type of Hereditary Punctate Palmoplantar Keratodermas J .Dermatology and Dermatitis. Doi:10.31579/2578-8949/003
Copyright: © 2017 Cesar Ricardo. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 06 February 2017 | Accepted: 22 February 2017 | Published: 26 February 2017
Keywords: punctate palmoplantar keratoderma; palmoplantar keratoderma; punctate keratoderma; buschke-fischer-brauer syndrome; acrokeratoelastoidosis; focal acral hyperkeratosis
Keratodermas encompass a wide spectrum of disorders of keratinization that may be acquired or hereditary. We present two cases of focal acral hyperkeratosis (FAH), a subtype of punctate palmoplantar keratoderma. We review the literature and attempt to clarify the confusing classification of the heritable punctate palmoplantar keratodermas.
Capsule Summary
1. (First bullet) what is already known on this topic
a. Punctate PPK is an esoteric topic within the field of dermatology that has been discussed both clinically and histologically.
2. (Second bullet) what this article adds to our knowledge
a. Despite this, the nosography and classification of punctate PPKs have been confused in published literature.
3. (Third bullet) How this information impacts clinical practice and/or changes patient care
a. This article hopes to clarify the terminology and classification of punctate PPKs, thereby increasing awareness of this disease, with the hope of advancing treatment options.
Palmoplantar keratodermas (PPK) may be categorized as acquired or hereditary. Acquired PPK have been associated with paraneoplastic syndromes and HIV/AIDS [1]. Inherited PPK are further classified by their distribution of epidermal involvement. The three classes of inherited PPKs are diffuse keratodermas, focal keratodermas and punctate keratodermas. These three classes of inherited PPKs can manifest in three ways; simple manifestation remains limited to the skin; complex manifestation includes lesions of nonvolar skin, hair, teeth, nails and sweat glands and syndromic manifestation can involve other organ systems, deafness, and/or cancer [2].
There are several types of punctate palmoplantar keratodermas (punctate PPK) described in the literature; however, classification is complicated by the misuse of names and redundant terminology. Type 1 punctate PPK, also known as Buschke-Fischer-Brauer disease, has been referred to in the literature as punctate PPK, lending to further confusion. Type 2 punctate PPK was referred to as porokeratosispunctata palmaris etplantaris; however, spiny keratoderma of the palms and soles is now favored (discussion to follow). Type 3 punctate PPK, or the marginal keratodermas, consist of focal acral hyperkeratosis and acrokeratoelastoidosis.
We present two cases of punctate PPK that were clinically and histologically consistent with the diagnosis of focal acral hyperkeratosis. In addition, the second patient presented with the rare association of FAH with keratosis punctata of the palmar creases. Recent literature demonstrates a renewed interest in these entities; however, ambiguous terminologies persist [3,4]. We therefore present an overview of hereditary punctate PPK, focusing on clarifying the terminology.
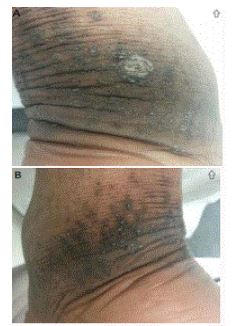
Punctate palmoplantar keratoderma. Multiple keratotic papules and crateiform pits overlying hyperpigmentation and lichenification traversing the Achilles tendon and limited to the right (1A) and left (1B) posterior ankles.
A healthy 29-year old African American woman presented to the clinic for evaluation of an asymptomatic lesions. The lesions were present for a number of years with a progressive worsening over this time. She received no previous treatment of the condition. The lesions remained localized to the areas of friction located over the posterior and posteriolateral ankles. The patient denied other skin concerns and had no past dermatologic history, including no history of atopic dermatitis or nail complaints. Of note, the patient reported that a sibling had similar lesions, but less severe invovement.
On physical examination, the patient was well-nourished. There were multiple papules with a central keratotic core and 2-5 mm circular depressions overlying hyperpigmented plaques, with mild lichenification located bilaterally on her posterior ankles, overlying the Achilles tendon (Figure 1a and b). Her palms and soles were spared and her fingers and toenails were normal in appearance.
A shave biopsy showed hypergranulosis and compact hyperkeratotic epidermis with focal overlying depressions. No columns of parakeratosis or elastorrhexis were noted (Figure 2a and b). These finding are consistent with punctate keratoderma [5]. Our patient was educated on pressure and friction reduction to the affected areas. Additionally, she was treated with keratolytics comprised of Urea 40% cream mixed with Tretinoin 0.025% cream applied twice daily to the affected areas. At follow-up 6 weeks later, there was significant improvement, with thinning of the lichenified plaques and spicules.
Case Presentation Two
A healthy 51 year old African American woman presented to clinic for presumed dyshidrotic eczema of the hands with secondary mild dyschromia that was initially treated with Fluocinonide 0.05% ointment (Lidex) twice daily and Ammonium Lactate 12% lotion (AmLactin) twice daily. The patient states that for years she had an increasing number of papules along the lateral aspects of her hands and fingers as well as a new mildly eczematous rash on her dorsal hands that improved with Lidex ointment. The patient denied other skin concerns and had no other past dermatologic history beyond skin xerosis. The patient reported that her father may have had similar skin findings.
On physical examination, the patient was well appearing. There were multiple umbilicated hyperpigmented papules bilaterally along the lateral aspect of the thenar and hypothenar eminences as well as pitted depressions on the creases of her palms. (Figures 3a and b) There were also multiple umbilicated hyperpigmented papules bilaterally in a linear like distribution, adjacent to the medial and lateral malleoli, sparing the soles (Figure 3c).
A shave biopsy showed compact hyperkeratosis with a focal depression (Figure 4a and b). An elastin stain was performed showing preservation of the elastin fibers (Figure 4c). The patient was started on Urea 40% cream once daily and was instructed to continue her previous regimen with Fluocinonide 0.05% ointment (Lidex) as needed for eczematous flares. She was also educated touse emollients liberally and to keep her hands dry and wear gloves whenever her hands are submerged in water. At followup 2 weeks later the patient noted diminished eczematous flares, with no other significant changes in her skin examination.

Punctate palmoplantar keratoderma. Skin biopsy revealed hypergranulosis with a compact hyperkeratotic epidermis and multiple epidermal depressions. Columns of parakeratosis and elastorhexis were not seen (A: Hematoxylin and eosin stain, original magnification x100, B: Hematoxylin and eosin stain, original magnification x200).
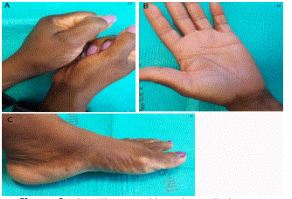
Punctate palmoplantar keratoderma. Multiple umbilicatedhyperpigmented papules bilaterally along the lateral aspect of the thenareminence (3A) associated with pitted depressions on the creases of the palms. (3B) and multiple umbilicatedhyperpigmented papulesadjacent to the medial malleoli, sparing the sole (3C).

Palmoplantar keratodermas (PPK) comprise a diverse group of inherited or acquired disorders that are characterized histologically by hyperkeratosis of the palmar and plantar skin. Punctate palmoplantar keratoderma (punctate PPK) is considered a hereditary subtype of PPK, so named for its presentation of multiple hyperkeratotic papules, spicules or nodules, classically found on the palms and soles in varying distributions. Genetic and environmental factors both play a role in the pathogenesis of punctate PPK. Keratodermas in general have been found with a greater prevalence in working class members (e.g. agriculturists), supporting the suggestion that the environmental influence of manual labor aggravates PPK [6]. Similarly, because of the continuous exposure to high pressure, lesions located on the soles of the feet can become confluent with time [7]. Several distinct subtypes of punctate PPK exist and are reviewed.
Type 1 punctate PPK, referred to by several other names (Table 2), most often occurs in adulthood but can range in onset from 12 to 30 years of age [8].The lesions have also been reported as presenting for the first time in the fifth decade of life [7]. It is characterized by multiple round or oval, firm and elevated hyperkeratotic papules that are yellow to dark brown in color, ranging from 2 - 9 mm in diameter [9]. The papules of Type 1 punctate PPK are diverse in appearance and may evolve with time, becoming translucent, opaque or verrucous in nature. Some papules may form a keratotic core and detachment of the core can result in a characteristic central depression [5]. These lesions are usually asymptomatic; however, patients may occasionally experience pain when pressure is applied to the affected areas [10].
Type 2 punctate PPK, erroneously referred to as porokeratosis of the palms and soles, has a distinct clinical manifestation of acuminata, or spiny, spike-like, keratoses in the palmoplantar region. When the filiform projections are not clinically obvious, spiny keratoderma of the palms and soles can be differentiated from other punctate keratodermas by histologic examination (Table 1). Onset of the lesions most often occurs between the second and fourth decade of life but case reports include ages ranging from 12 to 70 years old [11]. This disease entity is known for its confusing amalgam of terminology . The literature is increasingly advocating for adoption of the more accurate term “spiny keratoderma of the palms and soles” [12,13]. The accurate use of nomenclature is clinically important because in contrast to Type 2 punctate PPK, true porokeratosis can be associated with an increased risk of squamous cell carcinoma in the affected areas. Type 2 punctate PPK can easily be distinguished from porokeratosis both clinically and histologically. Histologically, Type 2 punctate PPK (spiny keratoderma) is composed of a column of parakeratotic cells, which is cornoid lamella-like, however it lacks the dyskeratotic and vacuolar changes that are found in true porokeratosis [11].
Type 3 punctate PPK consists of acrokeratoelastoidosis(AKE) and focal acral hyperkeratosis (FAH). AKE and FAH are also grouped to gether and referred to as marginal papularacrokeratodermas. AKE and FAH are clinically similar, presenting as 2-4 mm skin colored papules or plaques, uniquely presenting on the marginal borders of the hands, feet and/or digits [14]. AKE and FAH are inherited in an autosomal dominant fashion; however, sporadic cases have also been reported [15]. Lesions often present in the second or third decade of life, with one study reporting 87% of patients with symptoms before 20 years of age [16]. Lesions of AKE and FAH can appear umbilicated (crateriform) [17], often increasing in number and size over the years [18]. The distinction between AKE and FAH is made by the histological presence of elastorrhexis in AKE and its absence in FAH [17].
The lesions of AKE and FAH are typically asymptomatic but have been associated with hyperhidrosis in several case reports of AKE [19]. Both AKE and FAH may be associated with nail dystrophy [20,21]. Unusual reported variants of AKE/FAH include acquired crateriform hyperkeratotic papules of the lower limbs sparing the hands and feet [22] and acquired hyperkeratotic papules of the feet involving the dorsal and lateral aspects of the feet with sparing of the palms and soles [23].
Although many authors support the histologic distinction between AKE and FAH, others still debate the defining characteristics of these conditions [19,24]. The marginal papularacrokeratodermas (MPA) have had a complicated history of nosography. In 1994, Rongioletti et al. [14], proposed the division of MPA into hereditary and acquired forms, with further subdivision of the hereditary group based on the histologic presence or absence of elastorrhexis (Table 3). Among the hereditary MPA without elastorrhexis, they included FAH, acrokeratoelastoidosis of Matthews and Harman, hereditary papulotranslucent acrokeratoderma, and acrokeratoderma hereditarium punctatum. Mosaic acral hyperkeratosis was rescinded from this list by Rongioletti et al. [25]. The report concluded that the differences between these entities (hereditary MPA without elastorrhexis) should not be viewed as distinctive and should instead be considered variants of the same entity [14]. In 2000, Abulafia and Vignale disputed the classification of Rongioletti et al. [25], stating that the hereditary MPAs should not be considered as separate entities and are instead clinicopathologic variants of AKE [26]. Based on literature found in Pubmed under a search for “focal acral hyperkeratosis”, FAH has been adopted, with few dissentions, as its own disease entity with acknowledgement that it is clinically similar but histologically distinct from AKE. Despite the attempt to simplify and unify the nomenclature, the FAH variants reported by Rongioletti et al. [25], continue to be reported [3].
FAH, first named by Dowd et al. [17], was originally reported as a condition unique to the Afro-Caribbean population but it has since been found in Caucasians [14,19] and Asians [27,28,29]. However, a recent multicenter study performed in 2011 reported that a racial predominance of FAH in patients of African descent does exist. The study also noted that patients of African descent can have punctate keratosis limited to the creases of the palms, fingers and soles. This unique finding is referred to as keratosis punctata of the palmar creases, and is seen more often than FAH alone [30]. Furthermore, the coexistence of FAH with keratosis punctata of the palmar creases has rarely been reported in the literature [31,32]. Our second patient presents a case of FAH associated with keratosis punctate of the palmar creases.
The differential diagnosis of palmoplantar hyperkeratos is includes consideration of digitate keratoses of the palmoplantar surface, which includes spiny keratoderma, verrucae vulgaris and arsenical keratosis [13]. In contrast to Type 1 and Type 3 punctate PPK, digitate keratoses have a characteristic finding of minute fingerlike projections, descriptively referred to as “spiked,” “spiny,” “filiform” and so forth, or by the more imaginative name, “music box spines.” As discussed for spiny keratoderma of the palms and soles, the clinical distinction of spiny projections versus punctate papules is not always obvious. Furthermore, verrucae vulgaris and arsenical keratosis can manifest as both filiform and papular, making histologic examination essential for diagnosis. Verrucae vulgaris papules develop pinpoint bleeding when the lesions are pared [33]. Furthermore, dermoscopy reveals thrombosed capillaries in verrucae vulgaris, but not in punctate PPK. Arsenical keratoses may also appear clinically similar to punctate PPK, but a history of long-term arsenic exposure is required [34]. Arsenical keratoses lesions differ histologically with thick, compact hyperkeratosis and parakeratosis. Some epidermal keratinocytes may show atypia histologically. While the presence of numerous vacuolated keratinocytes and the absence of solar elastosis suggest arsenical keratoses, these findings are not definitive [35]. Important considerations in the differential diagnosis of palmoplantar pits also include nevoid basal cell carcinoma syndrome and plantar pitted keratolysis, hereditary and acquired disorders, respectively. Both disorders are easily distinguished from punctate PPK by histologic focal areas of absent or reduced stratum corneum [9].
Because it is an asymptomatic and benign condition, treatment of FAH is largely dependent on the patient’s desires and is often indicated for cosmetic reasons alone. A therapeutic goal in the treatment of FAH is to decrease hyperkeratosis with topical keratolytics such as urea, salicyclic acid and topical retinoids. Liquid nitrogen cryotherapy and prednisone have also been attempted, with poor results [36]. There is one report in the literature of acitretin successfully being used for treatment [29]. An earlier report published successful treatment of AKE with etretinate [37-39]. Our first patient responded to twice daily application of Urea 40% cream mixed with Tretinoin 0.025% cream, with significant improvement after 6 weeks. The second patient reported no significant improvement at 2 weeks follow-up with Urea 40% cream applied once daily. Longer follow-up of treatment for this patient is necessary to determine treatment efficacy.
Skin biopsy revealed compact acral hyperkeratosis with focal depression. The elastin stain showed preservation of the elastin fibers.
(A: Hematoxylin and eosin stain, original magnification x40, B: Hematoxylin and eosin stain, original magnification x200, Hematoxylin and eosin stain with EVG x200).
This reportpresents two cases of a unique type of hereditary punctate PPK known as FAH, or Type 3 punctate PPK. Owing to its asymptomatic nature, FAH has been underreported in the literature, therefore leading to a decreased awareness of its existence. With this case we hope to further elucidate an understanding of the classification of hereditary punctate PPK, thereby bringing greater awareness of FAH as a diagnostic potential. Furthermore, we propose inclusion of the Type 1A/B, 2 and 3 designations, as outlined in when referring to the various forms of hereditary punctate PPK. Although more cumbersome, it avoids ambiguity. It is our hope that a uniformly adopted terminology will facilitate further study and research of the punctate PPK group, thereby advancing our understanding of the disease and improving potential treatment options.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
