
Case Report | DOI: https://doi.org/10.31579/2641-0419/326
1 Department of Pediatric Cardiology, Klinikum Großhadern, Ludwig-Maximilians-University, Marchioninistraße 15, 81377 Munich, Germany.
2 Practice for Pediatric Cardiology, Auenstraße 6, 82467 Garmisch-Partenkirchen, Germany.
3 Department of Pediatrics, Dr. von Hauner Children's Hospital, University Hospital, LMU Munich, Germany.
4 Department of Pediatrics, Klinikum Garmisch Partenkirchen, Auenstraße 6, 82467 Garmisch Partenkirchen, Germany.
*Corresponding Author: Malin Zaddach, Department of Pediatric Cardiology, Klinikum Großhadern, Ludwig-Maximilians-University, Marchioninistraße 15, 81377 Munich, Germany.
Citation: Malin Zaddach, Julia Winter, Robert Dalla Pozza, Joachim J. von der Beek, Tobias Prell, et al, (2023), A Novel Ryanodine Receptor 2-Mutation associated with Catecholaminergic Polymorphic Ventricular Tachycardia, J. Clinical Cardiology and Cardiovascular Interventions, 6(6); DOI:10.31579/2641-0419/326
Copyright: © 2023, Malin Zaddach. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 31 May 2023 | Accepted: 12 June 2023 | Published: 05 July 2023
Keywords: schizophrenia; bipolar disorder; speckle tracking echocardiography; global longitudinal strain; cardiovascular disorders
Catecholaminergic polymorphicic ventricular tachycardia (CPVT) is a genetically determined arrhythmic disorder characterized by bidirectional and polymorphic dysrhythmias. Physical and/or emotional stress may trigger tachycardia, which can lead to sudden cardiac death. If not treated, CPVT has a high mortality rate of up to 31% by the age of 30 years. The prevalence of CPVT is estimated to be 1:10.000. Mutations of the ryanodine receptor 2, calsequestrin 2 and, less frequently, triadin and calmodulin are found in CPVT patients. We report the case of a four-year old boy with an unreported de novo mutation in the ryanodine receptor 2 (RYR2) confirmed by genetic sequencing. Interestingly, the first cardiac event of our patient was potentially be favoured by inhalative treatment with a short-acting beta-2 agonist.
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is characterized by adrenergic stimulus triggered arrhythmia [1]. The clinical diagnostic criteria include typical history, absence of structural abnormalities, normal resting ECG and provocation of polymorphic ventricular tachycardia under sympathetic stimulation [2,3]. Other typical ECG findings include bidirectional ventricular tachycardia [4]. Alternatively, diagnosis can be made by detection of a causative genetic variant. Correct diagnosis can be significantly delayed by misinterpretation of syncopes as seizures and learned avoidance behaviour of the patient [1,5]. Differential diagnoses include Brugada syndrome, Long QT syndrome, Short QT syndrome and arrhythmogenic right ventricular dysplasia. If not treated, CPVT has an extremely high mortality, which is 30-50% by the age of 40 years [6].
Mutations in six different genes have been identified to cause CPVT, which accounts for 60-75% of CPVT patients [7]. All currently known genes encode for proteins which are crucial for regulation of the Ca2+-induced Ca2+-release from the sarcoplasmic reticulum in cardiac myocytes. In 55-60% of CPVT cases, an autosomal-dominant inherited RYR2 (encoding ryanodine receptor calcium release channel) mutation is detected. Several proteins are known to interact with RyR2 to regulate calcium release (compare figure 3), including i.e. calmodulin, FKBP12.6, protein kinase A, junctin, triadin and calsequestrin. Mutations in CASQ2 (encoding for calsequestrin 2) are found in 2-5% of cases and can be inherited autosomal dominant and recessive. Rarely, mutations in TRDN (encoding triadin) and CALM1, CALM2 or CALM3 (encoding identical calmodulin proteins) are detected [7,8]. In all cases, Ca2+ homeostasis is disrupted and elevated intracellular Ca2+ levels lead to activation of the Na+/Ca2+ pump. In gain of function mutations delayed afterdepolarization, which may cause arrhythmias, is the consequence [8,9]. Loss-of-function mutations are also reported, promoting early afterdepolarizations and re-entrant arrhythmias [3]. In cases of loss-of-function mutations, exercise stress testing is negative and CPVT does present atypical and can be associated with left ventricular non-compaction [10]. Therefore, pharmaceutical therapy depends on either reduction of adrenergic receptor activation using β-blockers or modulation of calcium efflux from the sarcoplasmic reticulum using flecainide [11]. The article aims at the description of a clinical case, to discuss the pathophysiology related to the genetic findings, treatment options and influential factors.
Our patient is a 4-year-old boy without relevant previous diseases or long-term medication. The family history of sudden cardiac death was negative. The first cardiac event occurred while the boy had bronchitis for several days. Hence, the paediatrician had prescribed a short-acting beta-2 agonist (SABA, here: salbutamol) and budesonide inhalation. The mother left her son’s room for a moment right after he had inhaled. On her return, she found the boy lifeless and immediately started resuscitation. On arrival of the emergency physician, the ECG showed ventricular tachycardia (Figure 1) and a total of three defibrillations were performed before return of spontaneous circulation was achieved.

Figure 1: ECG on arrival of emergency physician. Left: Pre-defibrillation ECG showing ventricular tachycardia. Right: Post-defibrillation.
The patient was admitted to the hospital and extensive diagnostic workup was performed. Resting ECG showed normal sinus rhythm with QTc of 459 ms and incomplete right bundle branch block. Echocardiography did not reveal any structural anomalies and EEG did not show any epileptiform activity. Holter-ECG recorded a tendency to atrial extrasystoles (total of 3%), partially occurring in couplets. There was no ventricular extrasystole or tachycardia recorded. The patient was discharged with recommendation for further evaluation in a specialized referral center. An appointment was arranged for three weeks after discharge. Two days before the planned admission, an episode of fatigue, pallor and tachycardia prompted a further consultation at the emergency room. This time, recurrent ventricular extrasystoles were visible in the ECG on arrival and sinus rhythm was reconstituted after application of amiodarone (5 mg/kg of body weight). The patient was transferred to our department for further diagnostics, including electrophysiological study and cardiac magnetic resonance imaging. During electrophysiological investigation, a stress test using orciprenalin was performed revealing intermittent prolonged QTc to a maximum of 700 ms (amiodarone effect?). Therefore, Long QT syndrome was suspected. During continuous monitoring on our pediatric ward, occurrence of several arrhythmic events was noticed (Figure 2). Mostly, arrhythmogenic events precipitated by preceding stressing incidents (i.e. blood sampling, IV access, etc.), thus, offering the clinical possibility of CPVT. With the main suspicion of an ion channelopathy, genetic diagnostics was performed to identify the causative mutation. The patient received pharmaceutical therapy using propranolol with a dosage of 3 mg/kg of body weight [12]. No adverse effects were seen. On the last follow-up, the 50 h spanning Holter-ECG was graded 2 in the Lown grading system.
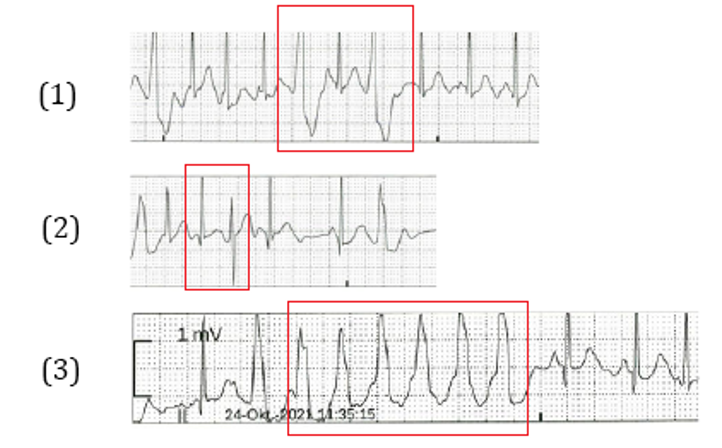
Figure 2: Examples for arrhythmogenic events: (1) Recurrent ventricular extrasystoles, (2) 180° turn of QRS-vector, (3) Ventricular salvo.
Trio whole exome sequencing (Illumina DNA Prep & TWIST comprehensive exome-enrichment with sequencing on NovaSeq 6000) revealed heterozygosity for a de novo Ile4094Val missense mutation, which had not been described elsewhere (GRCh37.p13: 1:237947292 A->G;
ENST00000366574: c.12280A>G). In silico analysis of our patient’s variant, using the algorithms Sorting Intolerant from Tolerant (SIFT) and Polymorphicism Phenotyping (PolyPhen), predicted a deleterious (0) and possibly damaging (0.809) effect concordant with a CADD Score of 25,6. In summary, the results of all applied bioinformatic prediction tools indicated a pathogenicity of the variant found.
We present a case of a 4-year-old patient presenting with sudden cardiac arrest due to ventricular tachycardia who was finally diagnosed with CPVT. Genetic testing revealed a novel Ile4094Val mutation in this case. The nonpolar, neutral amino acid isoleucine is substituted by the equally nonpolar, neutral, but smaller valine within the I-domain (interacting domain, amino acids 3722-4610) of RyR2, which has been characterized as a mutational hotspot (Figure 3). This region plays a key role in transducing regulatory cytoplasmic events to the C-terminal pore-forming transmembrane domain and mutations promoted abnormal Ca2+ release [4,8]. A variant in immediate vicinity to our patient’s mutation has been described as pathogenic before and bioinformatics analysis predicted a damaging effect underlining the pathogenicity of the identified mutation [13].
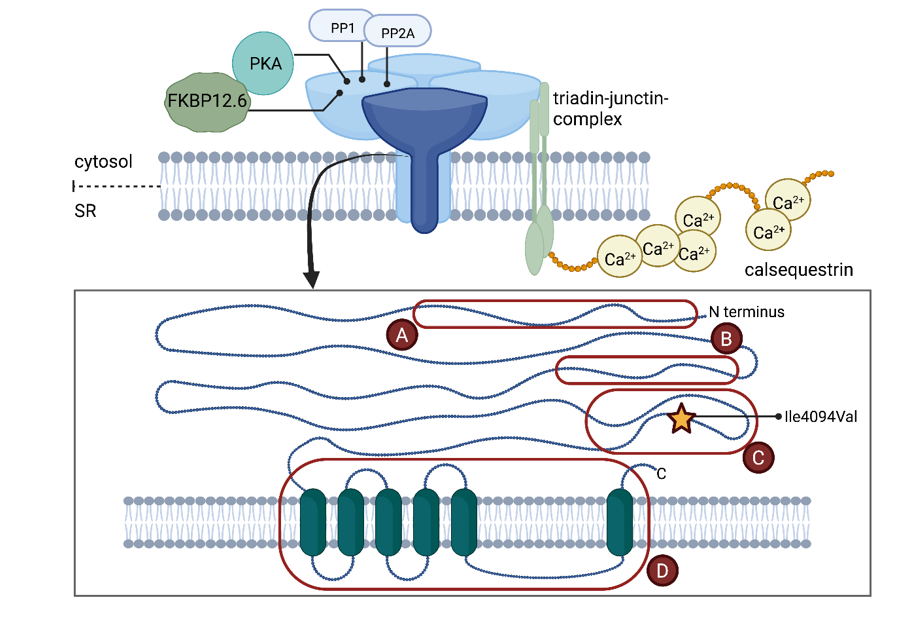
Figure 3: Schematic overview of the four subunits forming RyR2 and its interactions with protein kinase A (PKA), FKBP 12.6, protein phosphatase 1 (PP1), protein phosphatase 2A (PP2A) and triadin-junctin-complex, which interacts, in turn, with calsequestrin. Mutational hotspot regions of RyR2 are marked A-D. The variant found in our patient (Ile 4094Val) is highlighted with a yellow star. Figure created with BioRender.com.
Interestingly, the first cardiac event took place after inhalation of a SABA. Although, reports of arrhythmic events in connection with SABA inhalation are missing, there is evidence of a higher rate of supraventricular tachycardia in pediatric patients who underwent SABA treatment [14]. One large study reports an elevated risk of 1.89 (95% CI 1.31-2.73) of arrhythmia (mostly atrial tachyarrhythmia and premature ventricular and atrial beats) for children prescribed a SABA [15]. Seen the genetic predisposition of our patient, a interrelation between an increased vulnerability to arrhythmias triggered by beta agonist treatment seams possible.
RyR2 is not only found in cardiac myocytes but is also widely expressed in the brain, exerting a key role in Ca2+ homeostasis. RYR2 mutations were identified in some cases of benign epilepsy of childhood with centrotemporal spikes. The prevalence of intellectual disability is reported to be higher (8%) in CPVT patients as compared to the general population [16,17]. Interestingly, our patient showed mild cognitive delay and had been in occupational and speech therapy. Although the EEG was normal, we recommended further neurological follow-up for early recognition of epileptogenic activity.
Our patient was placed under appropriate medical therapy with β-blocker. Implantation of a cardioverter-defibrillator (ICD) was dispensed, as the events, which had occurred, had not been under pharmaceutical protection. In addition, occurrences of extrasystoles in physical or emotional stress situations were significantly reduced under monotherapy with propranolol. Complications such as infections, need for revision and non-adequate shock application are frequently seen in ICD-therapy, turning it into a second line therapy option in CPVT patients [18]. In selected cases medical therapy with propafenone, flecainide or carvediolol might be an option [19].
Although not obvious at the very first glance, the typical history of arrhythmic events provoked by inhalation of a sympathomimetic or emotional stress, a structural normal heart, normal resting ECG, a typical novel receptor mutation, which is located in the typically causative RyR2 protein, confirmed the final diagnosis of CPVT. In CPVT patients, a multidisciplinary approach on an individual basis including all therapeutic management strategies (i.e. medication, ICD, left cervical sympathetic denervation), is essential to achieve the best treatment possible, balancing safety and quality of life. [10,19]
Care-for-Rare Foundation, Munich
None.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
