
Case Report | DOI: https://doi.org/10.31579/2641-0419/371
Riojan Health Service, Emergency Service 061, Piqueras 98, 26006, Logroño, La Rioja, Spain.
*Corresponding Author: Alejandro Jesús Bermejo Valdés, Riojan Health Service, Emergency Service 061, Piqueras 98, 26006, Logroño, La Rioja, Spain.
Citation: Bermejo Valdés AJ, Jessica A. Jiménez, (2024), A Novel Presentation: Epsilon Waves in the Electrocardiogram of a Patient with Dravet Syndrome Without Structural Heart Disease, J Clinical Cardiology and Cardiovascular Interventions, 7(4); DOI: 10.31579/2641-0419/371
Copyright: © 2024, Alejandro Jesús Bermejo Valdés. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 12 April 2024 | Accepted: 08 May 2024 | Published: 16 May 2024
Keywords: dravet syndrome; epsilon waves; arrhythmogenic right ventricular dysplasia; sudden unexpected death in epilepsy; scn1a gene
We document for the first time epsilon waves on the electrocardiogram of a patient with Dravet syndrome (DS) who does not exhibit structural cardiac pathology. DS, a severe form of pediatric epilepsy, involves mutations in the SCN1A gene, which disrupt sodium ion flow and increase the risk of sudden unexpected death in epilepsy. The epsilon waves, typically associated with arrhythmogenic right ventricular dysplasia, may also suggest a link between neuronal and cardiac dysfunctions. In our case, the epsilon waves appear to originate from abnormalities in sodium channels rather than structural changes in the myocardium. This finding underscores the importance of assessing both neurological and cardiac manifestations in patients with severe epilepsy, opening new avenues for the research and treatment of these interactions in DS.
Dravet syndrome (DS) is a critical and severe type of pediatric epilepsy that not only presents with frequent and intense seizures but also has a notably high risk of sudden unexpected death in epilepsy (SUDEP). Despite extensive research, the exact biological mechanisms underlying the 15% incidence of SUDEP in DS remain elusive. [1-3]. The majority of DS patients have de novo mutations in the SCN1A gene [1,4,5]. These mutations disrupt normal sodium ion flow across neuronal membranes, affecting neuronal excitability and potentially contributing to the severe epileptic phenotype and increased SUDEP risk observed in these patients [1-5].
Ionic channelopathies are disorders affecting multiple organ systems, with significant implications across various physiological functions. Recent studies utilizing concurrent electroencephalogram and electrocardiogram (ECG) assessments have identified a notably high prevalence (33-44%) of arrhythmias in patients with epilepsy [6]. This data underscores the connection between neuronal and cardiac dysfunctions, suggesting that similar mechanistic pathways may be involved. Specifically, cardiac channelopathies can result from gain-of-function mutations, as observed in Long QT Syndrome Type 3, or from loss-of-function mutations, as demonstrated in Brugada Syndrome (BrS). Especially, BrS involves mutations in the SCN5A gene, responsible for encoding the myocardial sodium channel. This underscores the essential role of ion channel integrity in maintaining cardiac and neurological health [1,6].
In over 70% of individuals with DS, loss-of-function mutations are identified in the SCN1A gene [7-9], responsible for encoding the α subunit Nav1.1 of the voltage-gated sodium channel (VGSC) [4,5], present in both the brains and hearts of mammals. This genetic anomaly highlights the interconnected nature of “cardiocerebral channelopathies” where dysfunctions affect both cardiac and cerebral systems, potentially leading to sudden death [10].
Cardiocerebral channelopathies are, therefore, types of epilepsy that manifest with arrhythmias. Thus, epilepsies can be exhibited in various ways on the ECG. Specifically, we wish to highlight the presence of epsilon waves. These waves serve as a diagnostic criterion for arrhythmogenic right ventricular dysplasia, manifesting as delayed depolarization of this ventricle, with low-amplitude potentials located between the end of the QRS complex and the onset of the T-wave, particularly in the right precordial leads from V1 to V3 [11]. Although these waves can be detected in channelopathies involving sodium channels, such as in BrS [12,13], no specific cardiocerebral channelopathies involving these channels, such as DS, has ever been reported.
We present the first case of DS without structural cardiopathy, characterized by the presence of epsilon waves on the ECG
A 4-year-old child weighing 15 kg was attended at home by an Advanced Life Support Ambulance due to 16 generalized epileptic seizures in one day. The seizures are presumed to have been exacerbated by a fever of 39.5°C axillary, persisting for three days following vaccination. Upon physical examination, the patient was in an acceptable general condition. Pediatric Glasgow Coma Scale was 15. No neck stiffness or other meningeal signs were present. Cardiac auscultation revealed rhythmic heart sounds with a good tone at 160 beats per minute. The remainder of the examination showed no notable findings.
Regular treatment included: Valproic acid 28 mg/kg/day, Clobazam 0.1 mg/kg/day, Stiripentol 125 mg every 12 hours, Epidiolex 12.5 mg/kg/day, and Carnitine 300mg every 12 hours.
The patient’s medical history revealed the following:
In the electrocardiogram performed at the patient’s home, we observed a right bundle branch block of the His bundle and epsilon waves in the high left frontal leads DI and aVL, as well as in the right precordial lead V1. Figure 1 displays a recording at a speed of 50 mm/s with a vertical scale of 10 mm = 0.5 mV.
Figure 1: ECG recorded at 50 mm/s with a vertical scale of 10 mm = 0.5 mV. Epsilon waves are clearly visible in leads DI, aVL, and V1.At the hospital, the blood tests did not reveal any significant changes, except for mild neutrophilia with accompanying lymphopenia. Valproic acid levels were normal at 54 μg/mL (normal range: 50 – 100 μg/mL).
The SCN1A gene encodes the α-1 subunit of the Nav1.1, a critical protein that constitutes VGSC. Pathogenic variants cause a reduction in sodium GABA-ergic inhibitory interneurons, leading to hyperexcitability of the neuronal network and the onset of seizures [14,15]. SCN1A is associated with various epileptic syndromes and a range of other disorders [16,17].
Epsilon waves serve as essential diagnostic markers for arrhythmogenic right ventricular dysplasia, characterized by delayed depolarizations predominantly in the right precordial leads [11]. Should the left ventricle
be involved, epsilon waves can also manifest in the left and inferior leads [13].
The epsilon wave represents delayed potentials resulting from slow intraventricular conduction due to segments of normal myocardium interspersed with fatty and fibrous tissue. It corresponds to early afterdepolarizations once the QRS reaches the isoelectric line. Although the epsilon wave is a depolarization anomaly, it manifests on the ECG at the beginning of repolarization [13].
While epsilon waves are primarily associated with arrhythmogenic right ventricular dysplasia, they have also been documented in various other conditions unrelated to this disease. These include right ventricular myocardial infarction, Uhl’s anomaly (partial or complete absence of the right ventricular myocardium), and after repair of tetralogy of Fallot, where one case was noted to develop a right ventricular outflow tract aneurysm following episodes of ventricular tachycardia. Additionally, epsilon waves have been reported in cardiac sarcoidosis, sickle cell anemia in patients with right ventricular hypertrophy due to pulmonary hypertension, and BrS [12,13].
Generally, the presence of epsilon waves is linked to fibrosis or structural changes in the myocardium of the right ventricle [12]. However, an exception is BrS, which is primarily a cardiac channelopathy associated with sodium channels [1,6,18,19].
Two-dimensional Doppler echocardiography is highly sensitive to cardiac structural and functional changes and is used as one of the imaging tests in the diagnostic criteria for arrhythmogenic right ventricular dysplasia [20]. The absence of findings on this test in our patient rules out structural heart disease. This prompts the question of how to account for the presence of epsilon waves in our patient. We propose that the underlying mechanism behind the emergence of epsilon waves might be related to abnormalities in sodium channel function.
In the study conducted by Auerbach et al. [1], electrophysiological alterations were analyzed in a mouse model of DS through mutations in the SCN1A gene, which encodes the tetrodotoxin-sensitive Nav1.1 sodium channel. The results demonstrated a significant increase in both transient and persistent sodium current densities in isolated ventricular myocytes from these models. The researchers proposed that this increase could be attributed to augmented activity of the tetrodotoxin-resistant Nav1.5 sodium current, potentially resulting from a dysfunction in the Nav1.1 channel. This dysfunction could lead to a compensatory increase in the expression and functional activity of Nav1.5 channels in the membrane.
This heightened Nav1.5 current led to increased excitability, prolonged action potential duration, and the induction of early afterdepolarizations in the affected myocytes. Although the SCN5A gene encodes the α subunit of the Nav1.5 channel, which is predominant in mammalian hearts, it is important to note that Nav1.1 is also expressed in the ventricles, though its specific function in that region is still not clearly understood [1].
We propose that the presence of epsilon waves, not only in our patient but also in any patient with DS, could be attributed to early afterdepolarizations due to non-structural phenomena. We hypothesize that the involvement of sodium channels, expressed by the SCN1A gene at both cerebral and cardiac levels, may be the initial factor triggering an increase in Nav1.5-mediated sodium currents. Thus, we introduce the novel hypothesis that epsilon waves could originate from intrinsic molecular processes of the sodium channels, rather than simply being due to myocardial fibrosis or delays in right ventricular activation.
An outline of our reasoning up to this point is illustrated in Figure 2.
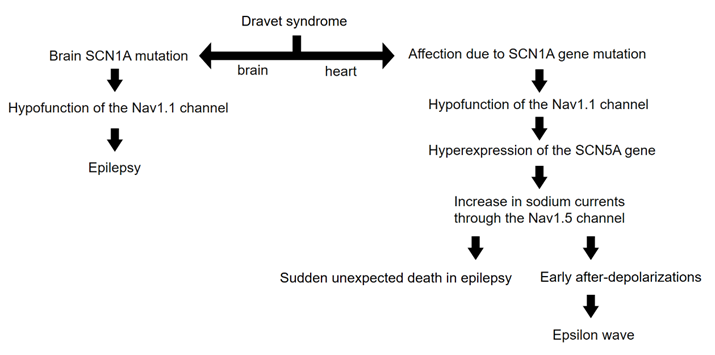
Figure 2: Diagram illustrating the reasoning behind the occurrence of epsilon waves in DS. Note that molecular and electrical phenomena are involved, rather than structural or anatomical phenomena. The basic mechanism would be the emergence of early afterdepolarizations due to the increase in cardiac sodium currents.
In our study, we have documented for the first time the presence of epsilon waves in the electrocardiogram of a patient with Dravet Syndrome who lacks structural cardiac pathology. These epsilon waves are crucial indicators for the diagnosis of arrhythmogenic right ventricular dysplasia and manifest as delayed depolarizations, typically associated with structural changes in the myocardium. However, in our case, the epsilon waves appear to arise from non-structural phenomena related to alterations in sodium channels.
We suggest that anomalies in sodium channels, particularly those associated with the SCN1A gene, could explain the emergence of epsilon waves in patients with Dravet Syndrome. Our findings propose an underlying mechanism where alterations in sodium channels, observed at both the cerebral and cardiac levels, might contribute to atypical electrocardiographic manifestations, including epsilon waves, without the need for myocardial fibrosis or structural changes.
Therefore, the implications of our study are significant for understanding the connections between epilepsies and cardiac arrhythmias, emphasizing the importance of considering both aspects in the clinical assessment of patients with severe epilepsy such as Dravet Syndrome. Furthermore, we highlight the need for further research to explore in greater depth the interactions between sodium channels in the brain and heart, which could open new avenues for therapeutic interventions addressing both seizures and cardiac complications in these patients.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
