
Research Article | DOI: https://doi.org/10.31579/2641-0419/219
1CSIR-Centre for Cellular and Molecular Biology, Hyderabad, India.
2Baba Clinical and Genomic Research Centre, CSIR Road, Taramani, Chennai, India.
3Academics and Research, Global Hospitals and Health City, Chennai, India
4Centre for DNA Fingerprinting and Diagnostics, Hyderabad, 500039, India.
*Corresponding Author: Deepa Selvi Rani, CSIR-Centre for Cellular and Molecular Biology Uppal Road, Hyderabad 500 007, India.
Citation: Deepa Selvi Rani, Gnana Veera Subhashini, Ambure Sharadhadevi, Emmanuel Cyril, Kumarasamy Thangaraj. (2021) A Missense Mutation (R723H) in the Head Motor Domain of β-MYH7 gene in an Indian HCM Patient and Phenotypic Plasticity. J. Clinical Cardiology and Cardiovascular Interventions, 4(17); Doi:10.31579/2641-0419/219
Copyright: © 2021 Deepa Selvi Rani, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 09 September 2021 | Accepted: 18 October 2021 | Published: 25 October 2021
Keywords: β-myh7; cardiomyopathy; homology models; 3d structure; sarcomere genes; hcm; dcm
Mutations in the β-MYH7 gene are one of the major causes that lead to cardiomyopathies. However, to differentiate a causative nsSNP and its impact on protein structure remains a major challenge. In the present study, we detected a missense mutation Arg723His in the head motor domain of β-MYH7 in a HCM patient, and it was absent in 207 healthy individuals. The mutant (R723H) has been found to alter an evolutionarily conserved amino acid. In addition, the mutant (R723H) was predicted pathogenic by Polyphen-2 and SIFT bioinformatic tools. Further, the superimposed 3D structure of the mutant (p.His723 homology model) with native (p. Arg723) displayed the root means square deviation (RMSD) of ~3.38A0. We know that the non-covalent interactions such as hydrophobic, electrostatic, Van der Waals, and hydrogen bonds between amino acids are at the heart of stabilizing protein structures. Here, we demonstrated how the mutant (p.His723) has disrupted a critical non-covalent interactions network at the mutation site and may contribute to the disease phenotype. Hence, our findings in the future could pave the way for developing small molecular modulators or myosin-targeted therapies for heart failure.
In general, non-synonymous Single Nucleotide Polymorphisms (nsSNPs) lead to change an amino acid of the encoded protein. They may affect protein structure, stability, and function, which may cause various diseases in humans [1-4]. About ≥50% of amino acids change are linked with genetic disorders [5,6]. However, a few amino acids change have remained uncharacterized in genes [7]. Mutations in the sarcomere proteins were reported to cause cardiomyopathies in various populations [8-15]. However, identifying the causative nsSNPs and their association led to disease is challenging. Cardiomyopathy is classified into Hypertrophic (HCM) and dilated (DCM) based on their heart muscle structure[16]. The HCM is characterized by excessive left ventricular thickening, arrhythmia, diastolic dysfunction, left ventricular outflow obstruction, myocardial ischemia, mitral regurgitation, and sudden death, with an estimated prevalence of 1:500[17]. Mutations in sarcomere genes predominantly cause HCM, of which ~75% of mutations were reported in the β-MYH7 and MYBPC3 genes [6,18-26]. Though the recent next-generation sequencing (NGS) technology has significantly increased our knowledge about disease alleles [27,28], we are far from completely understanding the impact of deleterious alleles on disease phenotype. We know that a few mutations may lead to a misfolding and nonfunctional form of proteins to accumulate and which may cause diseases. More importantly, the interactions between constituent amino acids in a protein determine its 3D structure and function [29]. Here, we have demonstrated a deleterious effect of a missense mutation (Arg723His) on β-MYH7 protein structure using (p.His723) homology modeling, an Insilco analysis.
Ethical statement and clinical evaluation
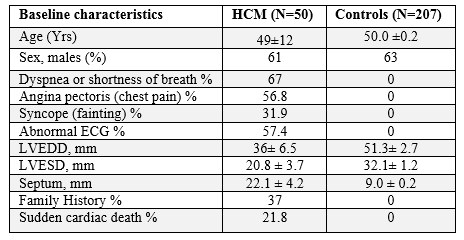
We enrolled a total of 50 hypertrophic cardiomyopathy patients (HCM) from 2 hospitals (Table 1). They were (a) Baba Clinical and Genomic Research Centre, CSIR Road, Taramani, Chennai, India, and (b) Academics and Research, Global Hospitals and Health City, Chennai, India. Along with 207 healthy volunteers matched for the age, sex and ethnicity were recruited as controls (Table 1), provided they had normal ECG and echocardiograph measurements and were unrelated to the HCM patients. The Institutional Ethical Committees (IEC) of all three institutes have approved the study. Before the sample collection, informed written consent was obtained from all patients and controls to fulfill the requirements of relevant guidelines and regulations that permitted research on human subjects, which has followed the ethics of the Declaration of Helsinki, the World Medical Association.
Genetic studies
The patients and controls DNA was extracted from peripheral blood samples, amplified using polymerase chain reaction (PCR), as described elsewhere [13]. The amplicons were purified using Exonuclease 1 and Shrimp alkaline phosphatase, following the manufacturer’s instructions (USB Corporation, 26, 111 Miles Road, Cleveland, Ohio 44128, USA). The purified amplicons were sequenced bi-directionally using the ABI Big Dye terminator cycle sequencing kit (Perkin–Elmer, Foster City, CA, USA) and ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA). Using Auto-Assembler software from Applied Biosystems (Foster City, CA, USA), the sequences were edited and screened for variations compared with the respective reference sequence obtained from Gen-Bank.
In silico analyses
A nonsynonymous single-nucleotide variant observed in our study was analysed using two bioinformatics tools, PolyPhen-2 (Polymorphism Phenotyping v2; http://genetics.bwh.harvard.edu/pph2/) [30] and SIFT (Sorting Intolerant From Tolerant; http://siftdna.org/www/Extended_SIFT_chr_coords_ submit.html).[31] Further, we built a homology model for a mutant of β-MYH7 by SWISS-MODEL Repository System (SMTL) (http://swissmodel.expasy.org) [32], using 3D native template structure having 99% similarity obtained
from the RCSB protein data bank (PDB) (http://www.rcsb.org/pdb/explore/explore.do?structureId=4P7H) [33]. To understand the impact of a nsSNP on β-MYH7 protein structure, we first superimposed the homology model of β-MYH7 with native β-MYH7 protein template structure to measure their root-mean-square deviations (RMSD) between the atoms (backbone atoms) of the superimposed pairs. We second studied the non-bonding interactions (created/destroyed) at the mutation site of the homology model vs. native β-MYH7. We then plotted the hydrophobicity plot and Ramachandran plot and studied the homology model vs. native β-MYH7.
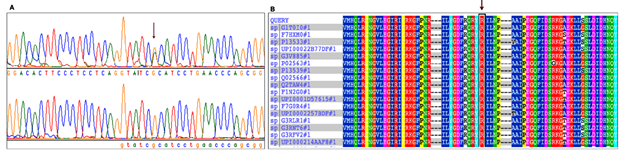
In the present study, we detected a missense mutation (R723H) in the head motor domain of β-MYH7 (Fig.1A). We found that the mutant (R723H) has altered the evolutionarily conserved amino acid across many species (Figure 1A, B). The mutant (p. His723) was predicted pathogenic by Polyphen2 and SIFT bioinformatics tools. Further, to understand the impact of mutant (Arg723His) on its protein structure, we first superimposed the mutant (p.His723_homology model) with native β-MYH7 protein (p.Arg723) and measured their root-mean-square deviation (RMSD), it was ~3.86Ao.
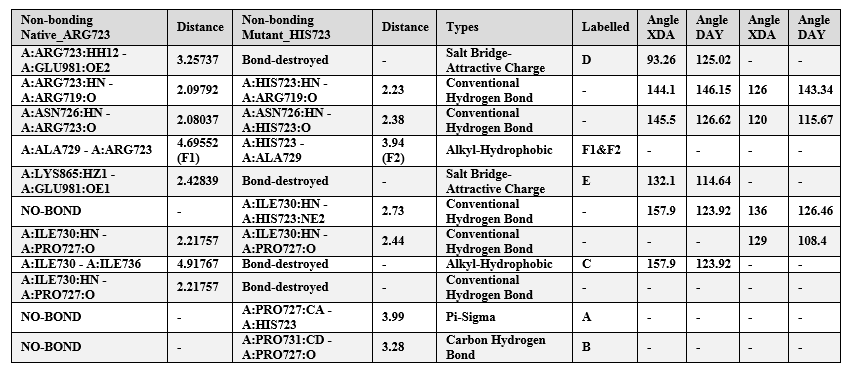
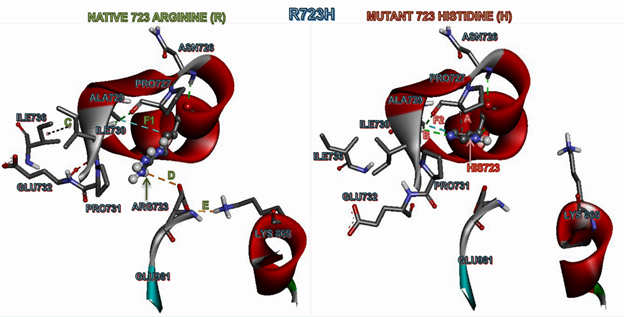
We then compared the non-bonding interactions of a homology model (p.His723) vs. native β-MYH7 protein to understand the mutational impact on protein structure and function (Table 2; Figure 2). Here, we observed that the mutant His723 forms a peculiar hydrophobic interaction with Pro727 (A), which, in turn, forms a hydrogen bond with another nearby proline residue Pro731 (B). As a result, a hydrophobic interaction between two isoleucine residues (Ile730 and Ile736) has been destroyed (C) due to an increased van der Waals radius (Table 2; Figure 2). Further, the mutant p.His723 also destroys two electrostatic salt bridges; Arg723 with Glu981 (D) and Glu981 with Lys865 (E) (Table 2; Figure 2). We know that the proline residue is unique and lacks an amide proton; therefore, it can't donate hydrogen to stabilize other bonds or promote stability, thus possibly making the mutant structure very rigid. The deviations in the mutant could be clearly understood when we compare the hydrophobic interaction distances between the native Arg723 with Ala729 (4.69 A0) in the template (F1) and the mutant His723 with Ala729 (3.93 A0) in the homology model (F2) (Table 2; Figure 2).
We plotted the hydrophobicity plot to compare the hydrophobicity index of native protein Vs. mutant protein (Figure 3; Table S1).
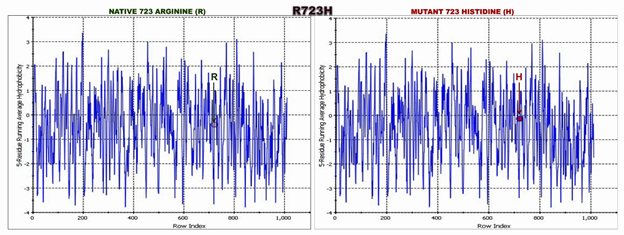
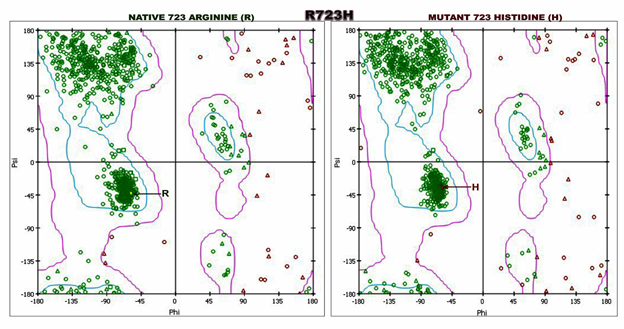
Mutations in a gene can be benign or pathogenic, and it is often challenging to establish which variants are pathogenic. The MYH7 is reported to be an important gene, and its mutations may lead to both HCM as well as DCM pathogenesis. Therefore, it is difficult to understand how a mutation can contribute to disease onset and how a gene mutation causes diverse cardiomyopathy phenotypes. In the present study, we found a nsSNP (Arg723His) in the head motor domain of β-MYH7 in a HCM patient (Figure.1A). However, we previously reported the same missense mutation Arg723His along with two other variations [(IVS19-1G) G>A, Ala729Ala] in exon 20 of the β-MYH7 gene (allelic heterogeneity) in a DCM patient and her son [34] [Rani et al 2021]. Thus, strongly reinforcing phenotypic plasticity in the presence of the compound mutation besides environmental background, epigenetic modifications/other factors, etc., [35,36]. The role of epidemiological factors in the pathogenetic process gains even more prominence because a single mutation can sometimes give rise to two very divergent phenotypes, emphasizing the role of gene modifiers and the influence of environmental factors in accounting for phenotypic plasticity [37,38]. This missense mutation R723H was absent in 207 healthy individuals. The mutant p.His723 has been found to alter evolutionarily conserved amino acids. It was predicted pathogenic by Polyphen-2 and SIFT bioinformatic tools. Further, we found that the mutant p. His723 (homology model) uniquely disrupts and deviates a critical network of non-bonding interactions at the mutation site (Table2; Fig.2). We know that a network of different kinds of non-covalent interactions between the amino acid residues drive the accurate 3D structure of the protein. Though different kinds of molecular interactions determine the accurate 3D structure of the protein, a network of non-covalent interactions between them is crucial [39]. We showed hydrophobicity plot (Figure.3; Table S1) and the Ramachandran plot (Figure.4; Table S2) to understand the deviation in mutant protein structure. Though different sequences map to a similar structure, a nsSNP can dramatically change a protein structure and lead to disease phenotype, such as sickle cell anemia (glutamic acid to valine (E6V) in the β-globin) [40]. Some studies suggest that the abnormal proteins themselves serve as pathogenic agents and are associated with various diseases [41]. However, functional studies are needed to confirm the actual pathogenic effect of this mutation.
Here, we have demonstrated how the (p.His723) mutant disrupted a critical non-covalent interactions network that possibly affects the structure and function. Therefore, understanding the impact of nsSNP on protein structure is indispensable for targeting the mutant amino acid residue for therapeutic purposes. Our findings in future could pave the way for developing small molecular modulators or myosin-targeted therapies for failing hearts.
We acknowledge all the hypertrophic cardiomyopathy patients and healthy controls for willingly contributed their blood samples for analyses.
Conflict of Interest
The authors have declared that there is no conflict of interest exist.
Funding Source
D. S. Rani is salaried through CSIR_CCMB, Hyderabad, India. CSIR/CCMB funded this project. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions
D.S.R. and K.T. conceived and designed the experiments. D.S.R performed PCR, Direct sequencing and mutation screening. D.S.R has done RMSD and Non-bonding interaction Insilico analysis with technical help from A.S. Cases and control samples were received from G.V.S., and E.C.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
