
Case Report | DOI: https://doi.org/10.31579/2690-4861/031
*Corresponding Author: Modupe Idowu, Division of Hematology, Department of Internal Medicine, The University of Texas McGovern Medical School at Houston, 6410 Fannin Street, Houston, Texas
Citation: Cinthia G, Amber C Goodspeed, Anam H, Modupe I. (2020) A Case of Macrophage Activation Syndrome Complicating Systemic-Onset Juvenile Idiopathic Arthritis. International Journal of Clinical Case Reports and Reviews. 3(1); DOI: 10.31579/2690-4861/031
Copyright: © 2020 Modupe Idowu, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 25 June 2020 | Accepted: 24 July 2020 | Published: 31 July 2020
Keywords: Macrophage activation syndrome; Juvenile Idiopathic Arthritis; hemophagocytic lymphohistiocytosis
Macrophage activation syndrome (MAS) is a rare but serious and potentially life-threatening complication of several chronic rheumatic diseases and can also occur in people with autoimmune or immunodeficiency conditions. It occurs most commonly with systemic-onset juvenile idiopathic arthritis, which is also known as Still's disease. MAS is thought to be closely related to and pathophysiologically very similar to hemophagocytic lymphohistiocytosis. A high index of suspicion should be maintained during diagnosis as correct early diagnosis is paramount to avoid serious complications. Here we present the case of a 23-year-old woman with a history of juvenile rheumatoid arthritis (JRA) who presented with a progressively worsening erythematous rash, cervical lymphadenopathy, splenomegaly, and pancytopenia. She was initially thought to be having a JRA flare, but she did not improve with steroids. She was then found to have MAS, which improved after treatment with cyclosporine. Although the duration of treatment of MAS with cyclosporine is not clear in the literature, we found that 3 months of therapy was sufficient.
Macrophage activation syndrome (MAS) is a macrophage-related histiocytic disorder [1]. It is a severe and potentially life-threatening complication of chronic rheumatic diseases most commonly seen in systemic-onset juvenile idiopathic arthritis (sJIA) [2]. Even though MAS can occur at any point during the course of sJIA, it typically occurs within the first few days or weeks of the onset of the disease [3].
Obvious MAS occurs in 10% of children with sJIA, but MAS can occur sub-clinically in another 30-40%, with some patients experiencing recurrent episodes [4]. The exact cause of MAS is unknown, but dysregulation of macrophage-lymphocyte interactions with a subsequent rise in the levels of both T cell- derived and macrophage-derived cytokines could be involved in this syndrome. This process is thought to lead to an intense systemic inflammatory reaction, which accounts for the main clinical picture of this disease. To successfully treat MAS, it is imperative to have a high index of suspicion, make an early diagnosis, and start prompt treatment [5].
A number of triggers have been linked to the development of MAS, including viral infections, especially Epstein-Barr virus infection, bacterial infection, parasitic infection, and fungal infection, and medications such as non-steroidal anti-inflammatory drugs (e.g., aspirin), anti-epileptic drugs (phenytoin, lamotrigine), methotrexate, sulfasalazine, gold salts, and anti-tumor necrosis factor-alpha drugs (etanercept, infliximab) [6]. In patients with sJIA, infections, flares of the underlying disease process, or changes in medication may trigger MAS [3, 7]. If MAS is triggered by an infectious agent or drugs, then the initial management will be the treatment of the specific infectious agent or withdrawal of the drug. However, MAS can occur without any identifiable precipitating factor [6].
Here we present the case of a 23-year-old woman with a history of juvenile rheumatoid arthritis (JRA) who presented with a progressively worsening erythematous rash, cervical lymphadenopathy, splenomegaly, and pancytopenia.
Case Presentation
A 23-year-old Caucasian woman presented to the Emergency Department with a 4-day history of lymphadenopathy of the right cervical chain and fever that were then followed by an erythematous rash. The rash started on the thighs and progressed proximally to involve the forearms, chest, and face. Her medical history was significant only for JRA, which was diagnosed at the age of seven, with her last flare being a year prior to this presentation. At the time of presentation, her only medications were tocilizumab, which she had been on for 2 years, and sulfasalazine, which was started by her rheumatologist a week prior. She had been taking sulfasalazine for 3 days prior to the onset of symptoms. On exam, she was found to have a blood pressure of 90/60, heart rate of 100 bpm, and oxygen saturation of 95% on room air. Her cardiac exam revealed normal S1 and S2 heart sounds without any murmurs, rubs, or gallops. Her lungs were clear to auscultation bilaterally. Her abdominal exam was significant for palpable splenomegaly. There were no focal neurologic deficits. There was no joint swelling or effusions present. There was a blanching maculopapular rash and pharyngeal inflammation without exudates.
The patient presented with an unclear clinical picture, but because of her history of JRA, the recent addition of sulfasalazine to her medication regimen, and her clinical presentation, the initial differential diagnosis included DRESS syndrome, JRA flare, sepsis, and MAS.
The patient’s initial labs showed an elevated alanine aminotransferase level of 75 U/L (normal 0- 65 U/L), aspartate aminotransferase level of 123 U/L (normal 0-37 U/L), lactate dehydrogenase (LDH) level of 764 U/L (normal 98-192 U/L), direct bilirubin level of 0.6 mg/dL (normal 0-0.3 mg/dL), prothrombin time international normalized ratio (INR) of 1.31 (normal 1.1) , and prothrombin time 16.5 seconds (normal 11-13.5 seconds). In addition, her labs were remarkable for a low platelet count of 69 K/cmm (normal 133-450 K/cmm), low fibrinogen of 182 mg/dL (normal 230-510 mg/dL), and an erythrocyte sedimentation rate of 1 mm/hr (normal 0-22 mm/hr) (Figure 1). A chest X-ray did not reveal any consolidation or infiltrates and a urine drug screen was negative. Other laboratory data include hemoglobin level of 13.9 g/dL, hematocrit of 39.8%, and white blood cell count (WBC) of 9.3 K/cmm (Figures 2, 3). However, even though the total WBC count was not elevated, on the differential, it was noted that the patient had 35% bands. There were no eosinophilia noted.
Given that sepsis was in the differential, the patient was not started on steroids on the day of admission. She had blood drawn for cultures and was started on broad-spectrum antibiotics. However, on day 2 of hospitalization, she was noted to have worsening liver function tests (LFTs), decreasing fibrinogen levels, increasing INR, and new-onset of shortness of breath. A chest x-ray was taken and revealed the development of pulmonary edema.

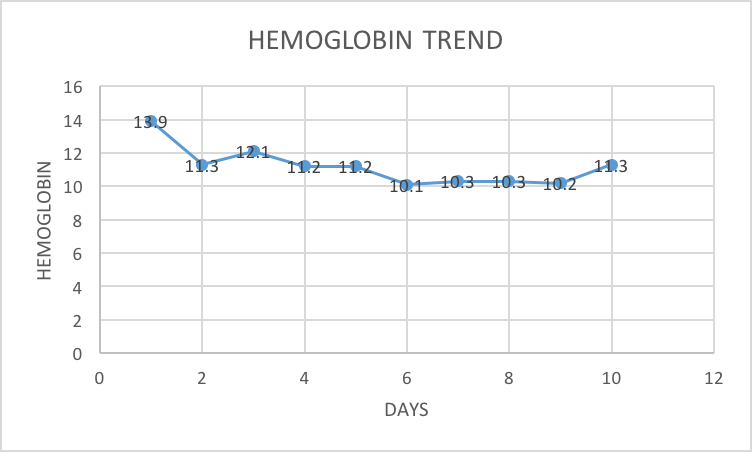

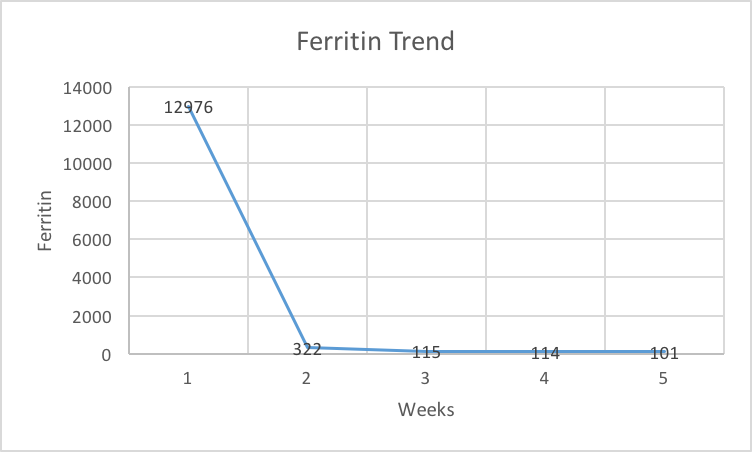
The worsening coagulopathy, transaminitis, and thrombocytopenia prompted a hematology consultation, which revealed that her platelet count had decreased to 54 K/cmm and her hemoglobin level had decreased from 13.9 to 11.3 g/dL (Figure 1, 2). Her LDH level had increased from 764 to 814 U/L, her total bilirubin level had increased from 1.3 to 1.9 mg/dL, her direct bilirubin level had increased from 0.6 to 1.2 mg/dL, and her INR had increased from 1.31 to 1.69. Her blood smear revealed a normocytic, normochromic anemia and reactive neutrophilia. There was no evidence of microangiopathic hemolysis. The hematology team initiated a further workup for MAS that included a lipid panel and ferritin level and scheduled a bone marrow biopsy. The ferritin level was found to be highly elevated at 12,976 ng/mL (normal 5 to 204 ng/dL), triglycerides were within normal limits, and high-density lipoprotein was low (Figure 4).
Treatment with steroids and cyclosporine was recommended if an immediate infectious workup was negative. The infectious workup included blood cultures, sputum cultures, and blood serology for cytomegalovirus, human immunodeficiency virus, Epstein-Barr virus, mumps, herpes, rubella, Pneumocystis jirovecii, and legionella. In addition, a bronchoalveolar lavage was performed because she had developed shortness of breath and pulmonary edema. Samples from the bronchoalveolar lavage were sent for viral culture, gram staining, and fungal culture, and tested for cytomegalovirus, influenza A and B, adenovirus, Pneumocystis jirovecii, and legionella. All infectious workup done on bronchoalveolar lavage samples was negative. All blood and sputum cultures were negative. A bone marrow biopsy was performed and showed normocellular marrow for age, absent iron stores, and rare macrophages with hemophagocytosis (Figure 5).
The combination of macrophages with hemophagocytosis on bone marrow biopsy and the laboratory findings of elevated LFTs, thrombocytopenia, decreased fibrinogen, elevated INR, and low erythrocyte sedimentation rate was suggestive of MAS (Figure 5). The patient was therefore started on cyclosporine 150 mg by mouth twice daily and dexamethasone 10 mg/m2 on day 6 of hospitalization. Sulfasalazine and tocilizumab were discontinued on admission as there have been reports of those agents triggering MAS [4].

The patient’s clinical condition as well as her laboratory findings dramatically improved after cyclosporine and dexamethasone were started. Her fever and shortness of breath resolved, and her platelet count increased from 45,000 to 103,000 per liter after 48 hours of cyclosporine initiation (Figure 3). Her transaminitis improved, with her aspartate aminotransferase level returning to normal and her alanine aminotransferase trending down from 123 to 118 U/L at the time of discharge. Her INR improved from its peak level of 1.69 to 1.1. She was discharged from the hospital day 10 on dexamethasone and cyclosporine, with a goal cyclosporine trough of 100-200. She was seen as an outpatient 1 week after discharge.
At discharge, her ferritin level was checked and had decreased from 12,976 on admission to 322 after being on cyclosporine and dexamethasone for only 2 weeks (Figure 4). Her lymphadenopathy and splenomegaly resolved within a month. Her cyclosporine dose was tapered slowly after 1 month of treatment, and her labs were followed every 2 weeks during the taper. Her dose was decreased from 150 mg twice daily, her initial dose for Month 1, to 100 mg twice daily for Month 2, followed by a change to 100 mg twice daily for 2 weeks, and then finally to 50 mg twice daily for 2 weeks. She therefore completed a total of 3 months of treatment with cyclosporine. Steroid was also tapered gradually over 1 month. She did very well after stopping cyclosporine, and her only complaint was that she noticed some hair loss, although she also observed that her hair grew during cyclosporine therapy. She has had no evidence of recurrence of MAS for 5 years.
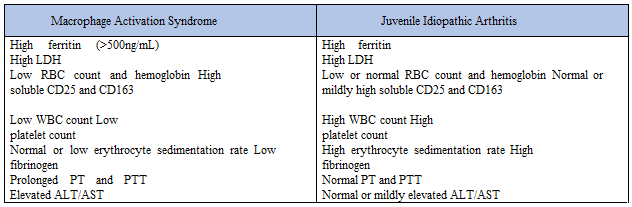
MAS is a rare but serious and potentially life-threatening complication of several chronic rheumatic diseases and can also occur in people with autoimmune or immunodeficiency conditions [1]. It occurs most commonly with sJIA. MAS is thought to be closely related to and pathophysiologically very similar to hemophagocytic lymphohistiocytosis (HLH). However, HLH criteria developed primarily for genetic disorders leading to hemophagocytosis are not necessarily useful in defining or identifying MAS. The main manifestations of MAS include fever, hepatosplenomegaly, hepatic dysfunction, lymphadenopathy, and disseminated intravascular coagulation. Moreover, patients may present with lethargy, seizures, coma, or shock. Unrelenting fever and rash, in contrast to the intermittent daily fever and rash of active sJIA, are also common findings. The white blood cell count, hemoglobin level, platelet count, and serum fibrinogen typically drop rapidly, while the ferritin level often rises much higher than 1000 ng/ml, similar to our patient’s clinical presentation (Table 1). There is a paradoxical drop in the erythrocyte sedimentation rate, as seen in our patient, due to fibrinogen consumption, which can be an important clue in the diagnosis of MAS [4]. On bone marrow examination of some patients, numerous benign macrophages exhibit hemophagocytosis, but not all bone marrow specimens from patients with MAS have this finding. MAS is difficult to diagnose in the setting of sJIA because systemic disease flare has a similar presentation; however, leukocytosis, neutrophilia, thrombocytosis, and elevated erythrocyte sedimentation rate are commonly seen in patients with sJIA without MAS.
Currently, there are no validated diagnostic criteria for MAS, as a result the 2004 HLH criteria being used to aid in the diagnosis of MAS [8]. However, the HLH diagnostic criteria cannot be strictly used for MAS in patients with sJIA despite the fact that the diseases are similar because the criteria are too stringent to identify MAS in sJIA patients early in the development of MAS when they are most likely to respond to treatment [9]. This difference in criteria is due to the fact that laboratory abnormalities, such as cytopenias, occur late in the MAS disease process, because patients with sJIA can have pre- existing neutrophilia and thrombocytosis due to their active inflammatory disease, which will hide decreases in these mature blood cells [1]. Following this reasoning, it is thought that the relative decrease in WBC count, platelets, or fibrinogen are better early diagnostic predictors of MAS than absolute deficiency is (Table 1) [10].
Currently there are no established treatment guidelines for MAS, but typically treatment is initiated with high-dose glucocorticoids (most often "pulse" methylprednisolone at 30 mg/kg, maximum dose 1 g, intravenously daily) [3, 4, 8]. Corticosteroids are used because they are not only cytotoxic to lymphocytes but also inhibit the production of CD95 ligand and the differentiation pathways of dendritic cells as well as the expression of chemokines and cytokines [11]. There are also case reports of successful treatment with cyclosporine or anakinra, an interleukin (IL)-1 receptor antagonist [11, 12, 13]. Cyclosporine A is a potent immunosuppressant that exerts its immunosuppressive action by interfering with the cyclophilin pathway. Its main effects are through suppression of the early steps in T-cell activation, leading to a failure to activate the transcription of “early” genes, such as those encoding for cytokines [14]. Cyclosporine A has also been shown to affect macrophage production of IL-6, IL-1, and tumor necrosis factor-alpha and to inhibit the expression of inducible nitric oxide synthetase and cyclooxygenase-2 in macrophages, leading to reduced production of nitric oxide and prostaglandin E2 [15,16]. Likewise, cyclosporine A has been proven able to inhibit the expression of key cell-surface co- stimulatory molecules, hence altering the antigen-presenting function of dendritic cells for T-cell activation [4].
For patients with rheumatologic disease who are diagnosed with MAS, treatment with increased immunosuppression and high-dose intravenous immunoglobulin are often effective. Biologic agents directed against inflammatory cytokines IL-1 and IL-6 have been used successfully in some patients [4, 17, 18, 19]. If the patient’s condition worsens despite steroids, cyclosporine, or other disease-specific therapy, it is sometimes necessary to intensify treatment to include etoposide, other HLH salvage therapy, or hematopoietic cell transplantation [1, 4, 8]. MAS is potentially fatal; therefore, timely diagnosis is critical. It is challenging to distinguish MAS from other conditions that may mimic some of its features, such as sJIA flares or sepsis‐like syndromes [20]. Differentiation of MAS from these conditions is crucial for selecting the appropriate therapeutic interventions in a timely manner.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
