
Case Report | DOI: https://doi.org/10.31579/2690-4861/010
*Corresponding Author: Jian Wang, State Key Laboratory of Respiratory Diseases, Guangzhou Institute of Respiratory Health, Guangzhou Institute of Respiratory Health, The First Affiliated Hospital of Guangzhou Medical University, Guangzhou, Guangdong, 510120, P.R. China.
Citation: Wang J, Jiang Q, Liu C, Wang C, Zheng R, Liu S, etc all (2020) A Case of Hemophagocytic Lymphohistiocytosis Coexisting with Pulmonary Hypertension. International Journal of Clinical Case Reports and Reviews. 2(2); DOI:10.31579/2690-4861/010
Copyright: © 2020 Jian Wang, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 30 November -0001 | Accepted: 26 February 2020 | Published: 04 March 2020
Keywords: pulmonary hypertension; hemophagocytic lymphohistiocytosis; hemophagocytosis; hematopoietic stem cell transplantation
Abstract:
Hemophagocytic lymphohistiocytosis (HLH) is a fatal disease associated with multiple organ dysfunctions and overwhelming inflammation. To date, pulmonary hypertension (PH) coexisting in patients with HLH is rare, and the treatment is unknown. Here we present an interesting case involving a 27-year-old male with dyspnea and irregular fever that eventually certified as PH coexisting with HLH. The patient finally died for severe infection 21-days post hematopoietic stem cell transplantation (HSCT). The necessity and timing of HSCT in this kind of patient need to be more careful.
A 27-year-old unmarried male with no prior history admitted to our hospital on 16/08/2017 with complaints of coughing and shortness of breath for 6 months, irregular fever for 1 month. Six months ago, he suffered from a worsening coughing without sputum and shortness of breath, more noticeable on climbing steep stair (no more than 20 steps). One month ago, he began to have a recurrent fever with maximum temperature of 39℃ , withoutchest pain, rash, nausea, vomiting, diarrhea, swollen lymph nodes, myalgias, decreased appetite and weight loss. He had no infectious disease, no allergies, no family history of heritable disease, and never used tobacco, alcohol or recreational drugs. He was a local food factory worker and denied occupational dust and radiation exposure. Physical Exam: His liver was palpated 5cm below the right costal margin and the spleen was 3cm below the left costal margin. No enlarged lymph nodes were palpated. He was first admitted to respiratory department of the Henan Provincial People's Hospital (22/06/2017-09/07/2017).
During his first hospitalization, echocardiography analysis suggested enlarged right heart (right ventricle (RV) 42.6 mm (normal range: 7-23 mm) and right atrium (RA) 55 mm (normal range: 33-41 mm)) and severe pulmonary hypertension (PH)with a pulmonary arterial systolic pressure (PASP) of 88mmHg (normal range: 15-30mmHg). The serum B-type natriuretic peptide (BNP) was 2773 pg/ml (normal range: <450 pg/ml). He was then diagnosed with idiopathic pulmonary arterial hypertension (IPAH) and initiated the therapy with sildenafil (25mg, tid) at 01/07/2017. His shortness of breath slightly improved after receiving the treatment and he was discharged on 09/07/2017. However, he still got a recurrent fever a few days after his discharge.
Then, he was re-admitted to our hospital on 16/08/2017. The computerized tomography pulmonary angiography (CTPA) demonstrated enlarged RV and RA due to PAH and no significant lesions in the lungs. Right heart catheterization (RHC) evidenced critical PH with a pulmonary arterial pressure of 37/14mmHg (mean 22mmHg), pulmonary capillary wedge pressure (PCWP) of 3 mmHg and pulmonary vascular resistance (PVR) of 3.8 Wood units (Table 1).

Moreover, echocardiography revealed an enlarged right heart (RV 42 mm and RA 46 mm) and moderately PH with a PASP of 60 mmHg. Consistent with RHC and echocardiography, BNP was mildly decreased in this patient (1382 pg/ml). V/Q scan, pulmonary function test, autoantibody (such as ANA, ANCA, ferritin, etc.), HIV antibody and portal venous pressure were at normal range. In conclusion, the diagnosis of IPAH was confirmed and the treatment of sildenafil was effective.
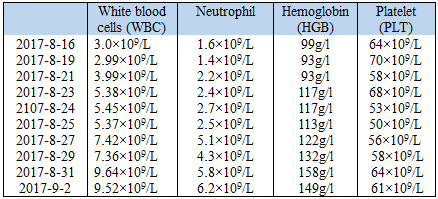
However, he still had a fever from time to time, even after broad-spectrum antibiotic (moxifloxacin, 400mg, qd). Besides, several things remain hard to explain based on the diagnosis of IPAH: the reduction of WBC, RBC and PLT in peripheral blood (Table 2), an ultrasound indicated hepatomegaly and splenomegaly (Figure 1) and elevated Epstein-Barr virus (EBV)-DNA (3.62*104 copies/ml in plasma).

A bone marrow biopsy was launched and revealed an increase of hemophagocytes (Figure 2).

In line with the finding of bone marrow biopsy, we detected a p.G863D (SNP rs140184929) and a p.K867E (SNP rs1135688) mutation in UNC13D gene, p.R49Q (SNP rs17073498) mutation in STX11 gene and p.I526V(SNP rs6791) mutation in STXBP2 gene (Table 3).

Moreover, the activity of natural killer (NK) cells was attenuated (the proportion of NK cells in lymphocyte was 2.99% (normal range: 7-40%)) and the serum level of triglyceride was raised to 2.16 mmol/L (normal range: 0.23-1.58 mmol/l). However, serum level of ferritin was normal (314.2ng/ml). Base on the HLH-2004 diagnostic criteria[1], this patient was then diagnosed as familial hemophagocytic lymphohistiocytosis (FHL), coexisting with PAH. He started to undergo two courses combination chemotherapy with etoposide (VP-16) (0.1g D1, D8) and dexamethasone (DXM) (10mg D1-D8) on 07/09/2017 and 19/09/2017. During this period, he was maintained on sildenafil (25mg, tid) and digoxin (0.125mg, qd) for the treatment of IPAH. Repeat bone marrow examination and echocardiography were performed on 27/09/2017, which showed remarkable decreases of hemophagocytes, decreased right heart (RV mm and RA mm) and PASP (36mmHg). Additionally, the patient's body temperature finally dropped to a normal range after chemotherapy (Figure 3).

Given the fact that ultimate treatment for FHLH is hematopoietic stem cell transplantation (HSCT)(1), he received allogeneic HSCT (allo-HSCT) on 10/10/2017, using HLA-half matched related donor (his mother). Unfortunately, he died for severe aspergillomycosis 21 days post-allo HSCT.
Discussion
Hemophagocytic lymphohistiocytosis (HLH) is a fatal disease associated with multiple organ dysfunctions and overwhelming inflammation. Classically, it is subdivided into two groups: (1) primary or familial HLH (also acknowledged as inherited HLH) and (2) secondary HLH[2]. Mutations in PRF1, UNC13D, STX11, STXBP2, RAB27A, LYST and SH2D1A genes have been identified as causative genetic defects of familial HLH. To date, it's still controversial whether EBV-driven HLH belongs to primary HLH[1,3]. Among these mutations,mutations of PRF1, Munc13-4, Syntaxin 11 and STXBP2 were then defined as FHL-2, FHL-3, FHL-4 and FHL-5, respectively [4,5]. And mutations in the genes of SH2D1A, BIRC4, ITK, CD27, and MAGT1 have been recognized in EBV-driven HLH [6] However, there was rare evidence reporting the genetic characteristics in Chinese adult patients with inherited HLH. In a study involving 26 Chinese pediatric primary HLH patients, the most common genetic defects were PRF1 (38.4%) and UNC13D (26.9%) [7]. In this case, we screened all the exons of PRF1, UNC13D, STX11, STXBP2, SH2D1A, XIAP, ITK, LYST, and RAB27A. Mutations in UNC13D, STX11 and STXBP2 were identified in this case. Although EBV was upregulated, EBV-associated genetic mutation was not found in this case. According to the diagnostic criteria of HLH-2004(1) we diagnosed this patient as familial HLH, which may be further classified as FHL-5. As the EBV-DNA was upregulated in this case, we also believed EBV infection may play a role in the disease process. And further studies focus on this issue need to be performed.
PH is a progressive, life-threatening disease, characterized by increased pulmonary arterial pressure [8]. In this case, not only repeated echocardiography confirmed increased PASP, but also CTPA and BNP evidenced the diagnosis of PH. However, RHC identified critical PH in this patient. The use of sildenafil (almost one month) before RHC examination may lead to this result. What is more, laboratory tests, such as negative autoantibody etc., also indicated the diagnosis of IPAH. Unfortunately, we did not perform a genetic test of IPAH in this patient. Notably, all the evidences suggested that both PH and familial HLH coexisted in this patient.
There are only few reports on PH coexisting in patients with HLH: Julius et al described a 64-year-old male with PH in the context of HLH [9]; Geraed et al reported a 52-year-old male with PH implicated in HLH [10]; Mitrovic et al. confirmed PAH was a complication of adult-onset Still’s disease [11] and Mili et al showed a 30-year-old woman with adult-onset Still’s disease complicated by both PH and macrophage activation syndrome (MAS) [12]. Moreover, PH was not a rare complication of HSCT for primary HLH [13,14]. Reviewing this case, whether patient with both PH and HLH benefited from HSCT was thought-provoking. And the timing of HSCT was also a challenge. To date, studies focus on this issue are rare. We recommend consultation of multi-disciplinary team (MDT) and close monitor of right heart function and PVSP before and after HSCT. Further studies should be performed to solve this problem. Besides, disseminated histoplasmosis and multiorgan failure were identified as etiological factors of secondary HLH [15,16,17]. The patient died for severe aspergillomycosis 21 days post-allo HSCT in our case, which reminds the awareness of fungal infection monitor should be strengthened.
Although the topic of PH involved in HLH has been illustrated, the underlying mechanism is still unknown. EBV was a well-known causative factor of both PH and HLH [18,19,20,21,22]. A retrospective analysis identified both HLH and PH were common complications for EBV infection [23]. PH was also observed in patient with chronic active EBV infection [18,21,23,24]. Furthermore, macrophage activation in HLH was also a potential mechanism for the development of PH [9].
There are limitations in this report: (1) the study of genetic defects of IPAH were absent here; (2) the lung pathology was lacked; (3) RHC should be performed before using sildenafil. HLH is a devastating disease, we should pay more attention if a PAH patient presents persistent fever together with the reduction of WBC, RBC and PLT in peripheral blood, hepatomegaly, splenomegaly and EBV infection.
Acknowledgments
This work was supported by the grants from the National Natural Science Foundation of China (81700048), Guangdong Department of Science and Technology Grants (2017A030310267), Independent Project of State Key Laboratory of Respiratory Disease (SKLRD-QN-201904).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Q. J., C. L., C. W. and R. Z. performed experiments; S. L. and Q. J. analyzed data; J. C. and J. Z. prepared figures; Q. J. and K.Y. drafted manuscript; N. Z., and J.W. conception and design of research; Q. J., K. Y., J. L, S. L. and J.W. edited and revised manuscript; J.W. approved final version of manuscript.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
