
Case Report | DOI: https://doi.org/10.31579/2690-1919/207
1 Department of Genetics, Children's Hospital, King Saud Medical City, Riyadh, Saudi Arabia
2 Resident – department of periodontology .King Saud Medical City, Riyadh, Saudi Arabia
3 Senior Orthodontic Resident, King Saud Medical City, Riyadh, Saudi Arabia
4 Medical Student, king Saud bin Abdul-Aziz University for health sciences
*Corresponding Author: Maha Alotaibi, Department of Genetics, Children's Hospital, King Saud Medical City, Riyadh, Saudi Arabia,
Citation: Maha Alotaibi, Deema Aldhubaiban, Ahmed Alasmari, Leena Alotaibi. (2021) A Case of Geroderma Osteodysplasticum Syndrome: Unique Clinical Findings. J Clinical Research and Reports, 9(2); DOI:10.31579/2690-1919/207
Copyright: © 2021 Maha Alotaibi. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 05 October 2021 | Accepted: 13 October 2021 | Published: 20 October 2021
Keywords: geroderma osteodysplasticum syndrome; lax skin; hyperextensible fingers; progeria; scoliosis; loose joints; abnormal hair
Geroderma osteodysplasticum (GO; MIM 231070) is characterized by a typical progeroid facial appearance, wrinkled, lax skin, joint laxity, skeletal abnormalities with variable degree of osteopenia, frequent fractures, scoliosis, bowed long bones, vertebral collapse, and hyperextensible fingers. The disorder results from mutations in the GORAB - golgin, RAB6 interacting. This gene encodes a member of the golgin family, a group of coiled-coil proteins on golgin. That maps to chromosome 1q24. The encoded protein has a function in the secretory pathway. Was identified by-teIrminal kinase-like protein, and thus it may function in mitosis? Mutations in this gene have been associated with geroderma osteodysplastica. Herein, we describe the clinical presentation of one young male patient from related Saudi parents. mutations, a homozygous Frameshift mutation (c.306dup p.(pro 103 Thrfs*20). Interestingly, phenotypic variability was observed in this patient with GO features that were atypical than the cases reported in the literature. As he looks tall stature where the most of cases reported were short and arachnodactyly of fingers which mimic and other syndromes.
Geroderma osteodysplastica (GO; OMIM 231070) is a rare autosomal recessive disorder of the connective tissue. Was delineated by Bamatter et al. [2] in five members of a Swiss family. That family has been reviewed for over 20 years [3,4] Those patients had facial dysmorphism, hyperlaxity of skin facies looks a droopy, prematurely aged appearance, The eyelids and cheeks droop. the forehead is prominent, the nose is often prominent and fleshy, maxillary hypoplasia osseous changes, variable severity of osteoporosis, hyperextensible joints, kyphoscoliosis, bone fractures and vertebral collapse, dental, CONGENITAL HIP dislocation. The disorder frequents in the Middle East and mainly in Oman. About 60 cases have been published to date [11].
The clinical phenotype of GO overlaps a heterogeneous group of disorders of the skin [5] including Cutis laxa syndromes, autosomal dominant cutis laxa (ADCL; MIM #123700), autosomal recessive cutis laxa (ARCLI; MIM #219100, ARCLII; MIM #219200, ARCLIII; MIM #219150), and wrinkly skin syndrome (WSS; MIM #278250) [6].
The GO disorder is due to the mutation of a gene that encodes for a Golgi apparatus protein called GORAB. The physiological function of GORAB is poorly defined. The golgin, RAB6-interacting (GORAB) protein localizes to the Golgi apparatus and interacts with the small GTPase RAB6 [7]. GORAB is important for vesicle transport at the Golgi complex and the correct processing of sugar chains on cargo proteins transiting through this compartment. Underlying defect of the skin and bone defects in GO patients is due to impaired COPI (coat protein complex I) trafficking at the Golgi apparatus resulting in abnormal glycosylation of extracellular matrix composition.
Clinical report
This ’A 24 year’ Saudi male (Figure 1) was born at term via breech vaginal delivery after limited antenatal care to a primigravida mother. He is a product of consanguineous healthy parents and the family history was negative for congenital malformations or deaths. He was seen initially in the dental clinic with generalized gingivitis gingival recession and severe crowding. (Figure 1) with dysmorphic facial features The facial features consisted of progeroid appearance, long triangular face deep-set eyes, droopy cheek, mid-face hypoplasia, mandibular prognathism, large prominent, a fleshy tip of the nose and prominent ears, hyperextensible fingers, arachnodactyly fingers and toes with the appearance of wrinkly skin, prominent veins in the dorsum of the hands., abdomen and joint laxity, His height was 187.6 cm and weight was 73.4 kg. Her upper: lower segment ratio was 0.81. Region. In addition, she had scoliosis with marked joint laxity. Radiological and skeletal findings support the diagnosis of GO Radiographs (Figure 2). DEXA scan showed a decrease in bone density (total lumbar spine (L1-L4) Z score -3.1; total right femur, Z score -2.5). No history of fracture and no recent hospitalization He is a college student with good performance, normal intelligence

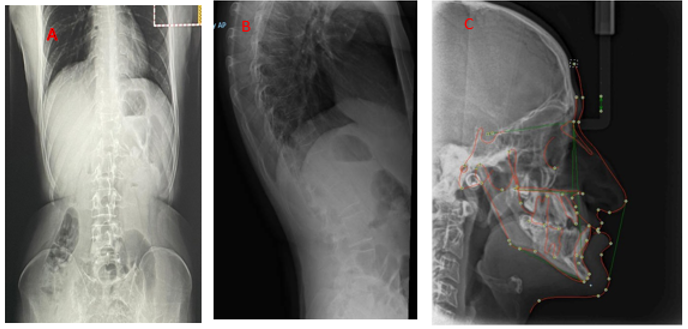
We report a GO patient with homozygous GORAB mutations with different clinical features that have not been reported before as tall stature and arachnodactyly and crowded teeth the unusual finding and distinctive clinical phenotype of GO emphasizes the usefulness of advance molecular genetic test for diagnosis.
The authors have no conflicts of interest
We highly appreciate the patient for consenting and participating in this study. The work is supported by the Department of Genetics, Children's Hospital, King Saud Medical City, Riyadh, Saudi Arabia.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
