
Case Report | DOI: https://doi.org/10.31579/2690-4861/225
* Department of Pediatric Nephrology, Turkey
*Corresponding Author: Deniz Karakaya, Department of Pediatric Nephrology, Turkey
Citation: Deniz Karakaya*, Yazılıtaş F., Evrim K. Çakıcı , Güngör T., Çelikkaya E., Bülbül.M. (2022) A Case of Adrenal Pheochromocytoma Presenting with Anxiety. International Journal of Clinical Case Reports and Reviews. 11(4); DOI: 10.31579/2690-4861/225
Copyright: © 2022 Deniz Karakaya, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 30 May 2022 | Accepted: 06 June 2022 | Published: 18 June 2022
Keywords: adrenal pheochromocytoma; neuroendocrine tumor; adrenal medulla
Pheochromocytoma is a rare, neuroendocrine tumor secreting catecholamine, arising from chromaffin cells of the adrenal medulla. Also, very rare cause of secondary hypertension in pediatric age, and however, it is important to suspect. Presentation of this tumor is highly variable but the most common pediatric cases being persistent hypertension, tachycardia, sweating, and headache. In addition, anxiety, weight loss and recurrent hypertension may also appear among the clinical manifestations of the disease. The diagnosis requires measurement of plasma metanephrines, with imaging studies performed for localization and identification of metastatic lesions. The definitive management of hypertension is surgical. We present a case with pheochromocytoma that was previously diagnosed as anxiety due to tachycardia associated with hypertension.
Hypertension (HT) is a high prevalent disease worldwide. The prevalence of pediatric HT approximately 0.3% to 4.5% and 5–10% of cases have a secondary cause which may be potentially curable [1]. So, accurate diagnosis of secondary HT is extremely important. Secondary HT is more common in children than in adults. This possibility should be excluded in childhood hypertension who presents symptoms, signs and laboratorial findings suggestive or specific for a secondary cause [1].
Pheochromocytoma (PCC) is a rare neuroendocrine tumor, secreting catecholamine, arising from chromaffin cells, originating from the adrenal medulla. It is also rare among the pediatric population; with an annual incidence of 0.2–0.3 cases per million and representing 9.6–17.7% of all cases [2,3].
The clinical presentation of this tumor is highly variable, as a result of excessive secretion of catecholamines; includes HT, tachycardia, sweating and headache. Hypertension may be continuous or paroxysmal. Orthostatic hypotension (low plasma volume and adrenaline effect), blurred vision, papilla edema, weight loss, polyuria, polydipsia, hyperglycemia, leukocytosis, symptoms such as psychiatric disorders are less common [4,5]. Treatment is surgical and includes stabilization of the patient's clinic until the time of surgery. We reported a boy who presented previously misdiagnosed as a psychiatric anxiety disorder with weight loss and palpitation complaints and two years later admitted with permanent HT.
A 15-year-old boy was admitted to the outpatient clinic of Pediatric Nephrology at Dr. Sami Ulus Maternity and Children's Health and Diseases Training and Research Hospital for hypertension accompanied by headache, epigastric pain and weight gain.
Two years ago, his blood pressure was high when he was evaluated with similar complaints. He was diagnosed as anxiety disorder because of occasionally observed high blood pressure, tachycardia, headache and feeling nervous. He had received short-term anxiolytic treatment at that time. Then he discontinued his medication because his complaints sustained. Also, he was followed up in an eye center for macular edema for two years. The patient has not been taking any medications for the past year and there was no smoking use or alcohol consumption. He had no family history of any disease and HT.
On admission he had marked high blood pressure (170/110 mmHg) with 2/6 grade systolic murmur in mesocardiac area. Physical examination showed body mass index of 22.8 kg/m2, heart rate was 78 beats per minute-rhythmic and no fever, abdomen murmurs or palpable masses and edema. His neurological examination was normal. Funduscopic examination revealed swollen optic discs and macular star appearance. He had a normal complete blood count and normal renal function and hepatic enzymes. Serum cholesterol and triglyceride levels were normal. There was no proteinuria in urine analysis.
An aortic insufficiency and left ventricular hypertrophy was detected in echocardiography and ejection fraction (70%) was normal. Abdominal ultrasound (US) revealed a hypoechoic adrenal mass measuring 50 x 47 mm in size in the paravertebral area. Magnetic resonance imaging (MRI) showed a 65x45 mm hyper vascular lesion with a central necrotic region on the left adrenal gland, compatible with a PCC [Figure 1 and 2]. No extra-adrenal tumors were seen.
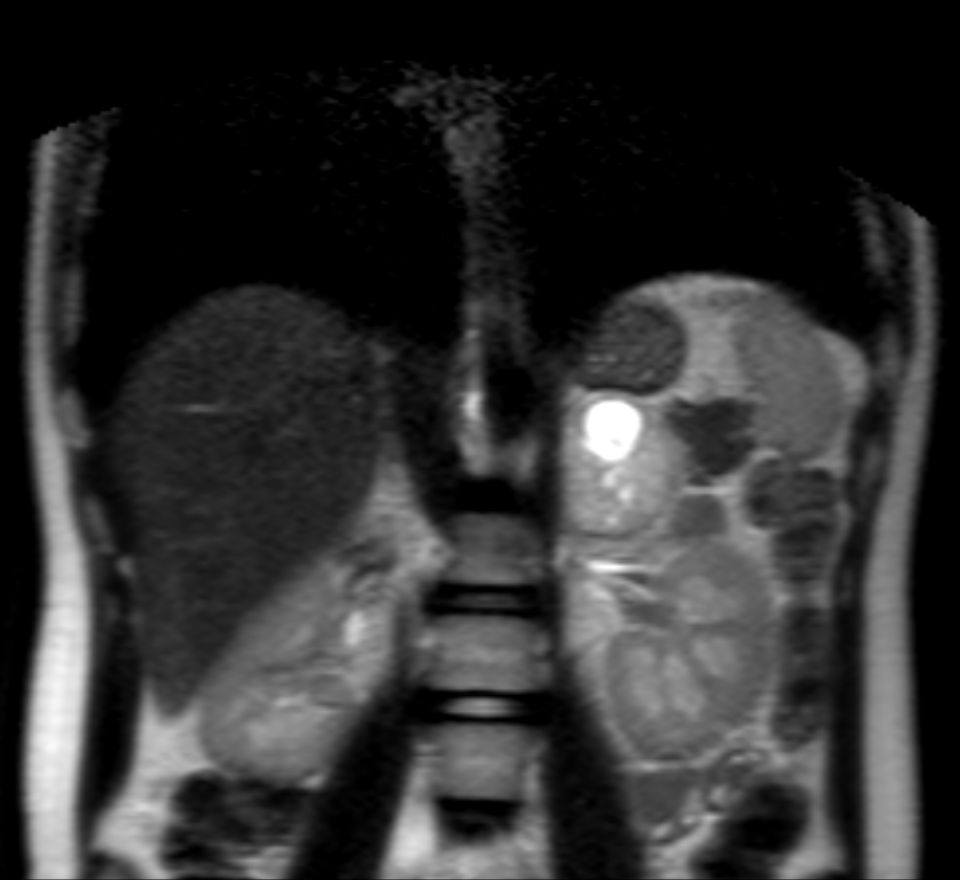
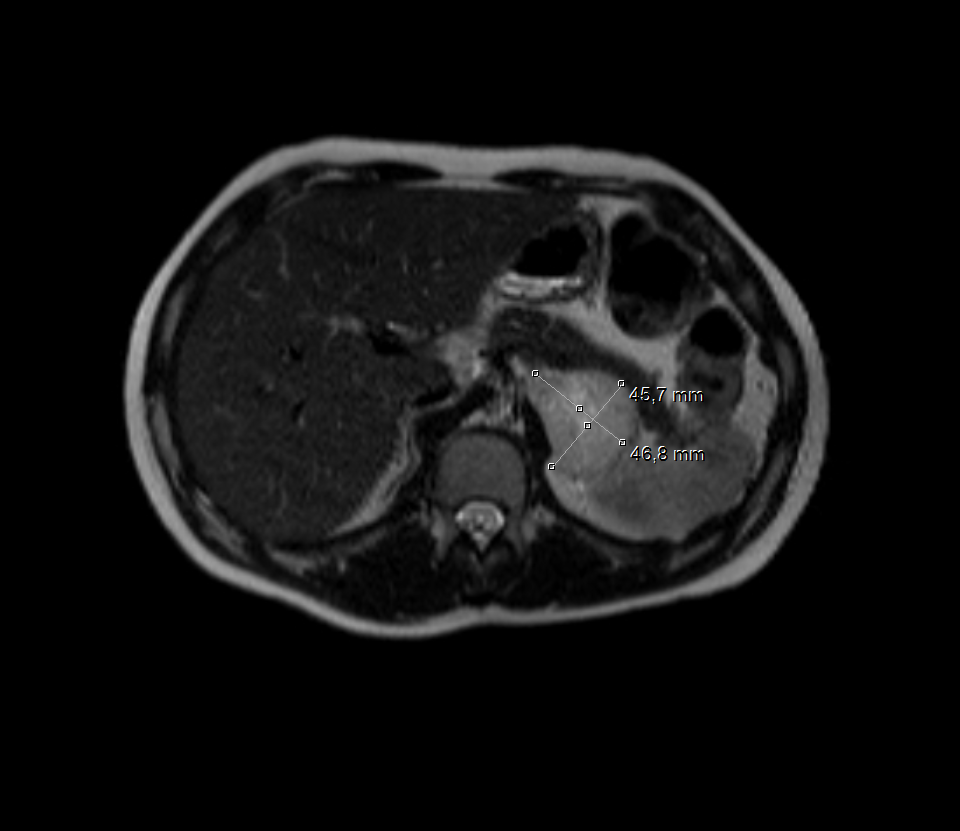
Thyroid and parathyroid ultrasound were normal. His other investigations were: blood renin 10.12 ng/ml/h (0.5-5 ng/ml/h), blood neuron-specific enolase 25.6 ng/mL (0-17 ng/mL), spot urine metanephrine 170.95 mcg/g creatinine (30-154 mcg/g), and urine metanephrine 254.59 µg/24h (52-341 µg/24h).
Alpha-adrenergic blocker, calcium channel blocker, angiotensin converting enzyme (ACE) inhibitor and beta blocker were given for blood pressure control. The patient's blood pressure was controlled during follow-up. Surgical intervention was recommended for further treatment and he was referred to an advanced center for surgical intervention. The patient had normal blood pressure control before and during the operation. There was no complication. Postoperatively, the patient was re-admitted to follow-up. His blood pressure was normal without anti-hypertensive medicine. The levels of 24-h urinary metanephrines and normetanephrines were normal. At the three-month follow-up, the patient's weight gain was approximately 10 kilograms.
In the present study, we reported a patient with pheochromocytoma, who was previously diagnosed as a psychiatric anxiety disorder due to presence of weight loss and tachycardia symptoms and HT.
Hypertension is an important health problem worldwide, is becoming widespread in childhood and may cause important complications. Various risk factors have been associated with pediatric HT. Secondary HT is more common in younger children resulting from reno-vascular or renal parenchymal disease (78–80% of causes), endocrine (11% of causes), cardiac (2% of causes), pulmonary and others [6,7,8]. Pheochromocytoma accounts for only 0.5-2% of secondary HT causes (6). Since Although PCC is the cause of very few secondary HT, it is a disease that must be kept in mind when evaluating a child with hypertension.
Pheochromocytoma, a rare chromaffin cell tumor, secretes catecholamine which is responsible for the symptoms such as weight loss, tachycardia, sweating, HT, headache and tachycardia. Also, orthostatic hypotension (low plasma volume and adrenaline effect), blurred vision, papilla edema, weight loss, polyuria, polydipsia, hyperglycemia, leukocytosis, symptoms such as psychiatric disorders are less common [4,5]. It is known that most of the pediatric patients have HT and most of them have continuous hypertension rather than paroxysmal HT as in our case. HT is associated with various complications. Retinopathy in the eye secondary to HT [9] and left ventricular hypertrophy [10] in the heart were observed in our patient. In our case; HT, weight loss and palpitations, which occurred two years ago, caused to be diagnosed with anxiety. Also, anxiety is one of the rare psychological findings of PCC [11]. Although not described as a typical symptom of PCC, anxiety is the fourth most common symptom reported by patients suffering of PCC (11).
Diagnosis is made by demonstrating increased urinary and plasma catecholamine and metabolites. Metanephrines increased in 95-97% of patients. Plasma metanephrine and normetanephrine levels are very high negative predictive values. Normal plasma concentrations eliminate the diagnosis of PCC [12]. In our patient, urinary methanephrine and blood neuron-specific enolase levels were found to be high consistent with and the diagnosis of PCC. After biochemical diagnosis of PCC, the next step is to detect the tumor, radiological imaging studies. Contrast enhanced computed tomography is recommended as the first-line imaging modality for evaluation with a reported sensitivity of 88–100%, while MRI has been recommended for detection of head and neck paragangliomas (PGL) [13], and positron emission tomography for metastatic disease [14,15]. It is necessary to have both anatomic and functional imaging when evaluating all pediatric patients who present with PCC/PGL. In our case, MRI was preferred in the second stage to evaluate the mass.
The definitive treatment of PCC is surgical removal of the tumor. The patient must be stabilized before the operation; blood pressure should be normal for at least 1-2 weeks and no arrhythmia. Medical treatment should be started when suspected about disease. The aim of treatment is to reduce symptoms, lower blood pressure, and prepare patients for surgery after localization of the tumor. The first choice in controlling blood pressure is phenoxybenzamine, a long-acting adrenergic blocking drug [16]. Alpha-blockade is the standard management preoperatively to prevent intraoperative hemodynamic instability during resection of a mass [16]. Pre-operative and postoperative well controlled blood pressure should be ensured because catecholamine can be discharged during operation. Recurrence or persistent hypertension may be seen in 25% of the cases [17]. It has not been detected in the follow-up of our case. Also urine or plasma catecholamines and their metabolites should be measured at two weeks postoperatively. The measurements are repeated every three months in the first year, and then are repeated every 6 or 12 months in follow-up [17]. Imaging should be performed after a few months in the postoperative period and then as often as required [17].
In conclusion, although catecholamine secreting tumors originating from chromaffin cells are rare, they should be considered in childhood that observed HT. Pheochromocytoma in children may present with different symptoms such as anxiety. Childhood HT should be investigated carefully before it is thought to be associated with psychological factors such as anxiety.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
