AUCTORES
Globalize your Research

Case Report | DOI: https://doi.org/10.31579/2641-0419/463
Department of Cardiac Anaesthesia, Medanta-The Medicity, Gurgaon (Haryana)-122001, India.
*Corresponding Author: Ajmer Singh, Director, Cardiac Anaesthesia Medanta-The Medicity, Gurgaon (Haryana)-122001, India.
Citation: Ajmer Singh, Ravina Mukati, Ruchi Agrawal, (2025), Quadricuspid Aortic Valve: Intraoperative Diagnosis by Transesophageal Echocardiography, J Clinical Cardiology and Cardiovascular Interventions, 8(6); DOI:10.31579/2641-0419/463
Copyright: © 2025, Ajmer Singh. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 21 March 2025 | Accepted: 09 April 2025 | Published: 18 April 2025
Keywords: quadricuspid aortic valve; aortic regurgitation; aortic valve replacement, transesophageal echocardiography
The quadricuspid aortic valve is a rare congenital anomaly, which usually progresses to aortic regurgitation in adulthood requiring aortic valve replacement. It may be associated with other congenital abnormalities such as aberrant coronary arteries, patent ductus arteriosus, ventricular septal defect, pulmonary stenosis, persistent left superior vena cava, subaortic stenosis, and aortic dilation. Most cases are diagnosed incidentally during aortic valve surgery or autopsy. With the availability of better imaging techniques such as three-dimensional echocardiography and cardiac magnetic resonance imaging, more cases are being diagnosed in asymptomatic patients. Herein, we report a case of a 55-year-old man in whom a quadricuspid aortic valve was detected by intraoperative transesophageal echocardiography.
Quadricuspid aortic valve (QAV) is a rare congenital malformation with an estimated incidence of 0.008% to 0.043%.1 Most cases are discovered incidentally during aortic valve surgery or autopsy. The condition frequently progresses to aortic regurgitation in 75% of the patients, which generally manifests in the fifth decade of life with a mean age of 50.7 years, and may require surgical treatment. Combined aortic regurgitation and stenosis affect only 8% of the valves and normal functioning is seen in about 16% of patients.2 QAV may be associated with other conditions, including abnormalities of other cardiac valves, coronary anomalies, septal defects, persistent left superior vena cava, patent ductus arteriosus, pulmonary stenosis, and aortic dilation in one-third of the patients.
There are two classification systems for QAV: first the Hurwitz and Roberts classification and second the Nakamura et al classification.3,4 In the former, the QAV is classified according to the size of the supernumerary or accessory cusp into seven categories from A to G. According to this classification system, type A contains 4 equal-sized cusps; type B, 3 equal cusps and 1 small cusp; type C, 2 equal large and 2 equal small cusps; type D, 1 large, 2 intermediate, and 1 small cusp; type E, 3 equal and 1 larger cusp; type F, 2 equal larger and 2 unequal smaller cusps; and type G, 4 unequal cusps. Type A and type B are the most common variants, but it is unclear whether any specific subtype of QAV predisposes the patient to more severe aortic regurgitation. The classification system proposed by Nakamura et al is based on the position of the accessory cusp, whether it is located between the right and left coronary cusps (type 1), between the right and non-coronary cusps (type 2), between left and non-coronary cusps (type 3), and type 4 when there are four equal sized cusps present.
A 55‑year‑old man with progressive dyspnea on exertion (New York Heart Association functional class III), fatigue, and swelling of feet was admitted for evaluation. Physical examination showed bounding pulses, early diastolic murmur on the left lower sternal border, S3 gallop rhythm, and a blood pressure of 128/42 mm Hg. Chest radiograph showed cardiomegaly. Preoperative transthoracic two-dimensional echocardiography showed a trileaflet aortic valve, severe aortic regurgitation, and severe left ventricular dysfunction. After obtaining informed consent, the patient was taken up for aortic valve replacement. Intraoperative transesophageal echocardiography (TEE) showed a QAV (mid‑esophageal aortic valve short‑axis view) with a rudimentary accessory cusp located between the right and non-coronary cusp (Figure 1). There was moderate aortic regurgitation, left ventricular dilation (end-diastolic dimension 5.7 cm), and left ventricular dysfunction (Figure 2). An en-face view with three-dimensional (3-D) echocardiography confirmed the TEE findings (Figure 3). The identification of four cusps was facilitated by placing 4-D markers on individual cusps. The surgical findings showed three large cusps and one smaller cusp located between the right and non-coronary cusps (Figure 4) making it type ‘C’ QAV (Hurwitz and Roberts classification). Under Nakamura’s classification, it was type 2 QAV with the accessory cusp located between the right and non-coronary cusp. The other cardiac structures including the valves and ascending aorta were normal. After excision of the native aortic valve, the patient underwent aortic valve replacement with a mechanical prosthesis uneventfully. Further course of the patient in the hospital was unremarkable.
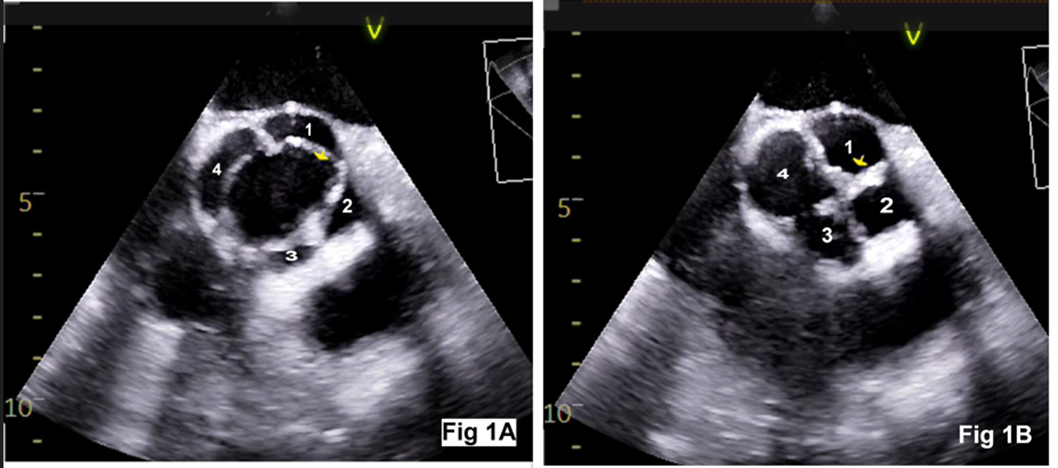
Figure 1: Transesophageal echocardiography aortic valve short-axis view showing rectangular opening during systole (Figure 1A) and ‘X’ shaped closure during diastole (Figure 1B). The cusp numbers denote: 1-left coronary cusp, 2-right coronary cusp, 3-accessory cusp, 4-non-coronary cusp
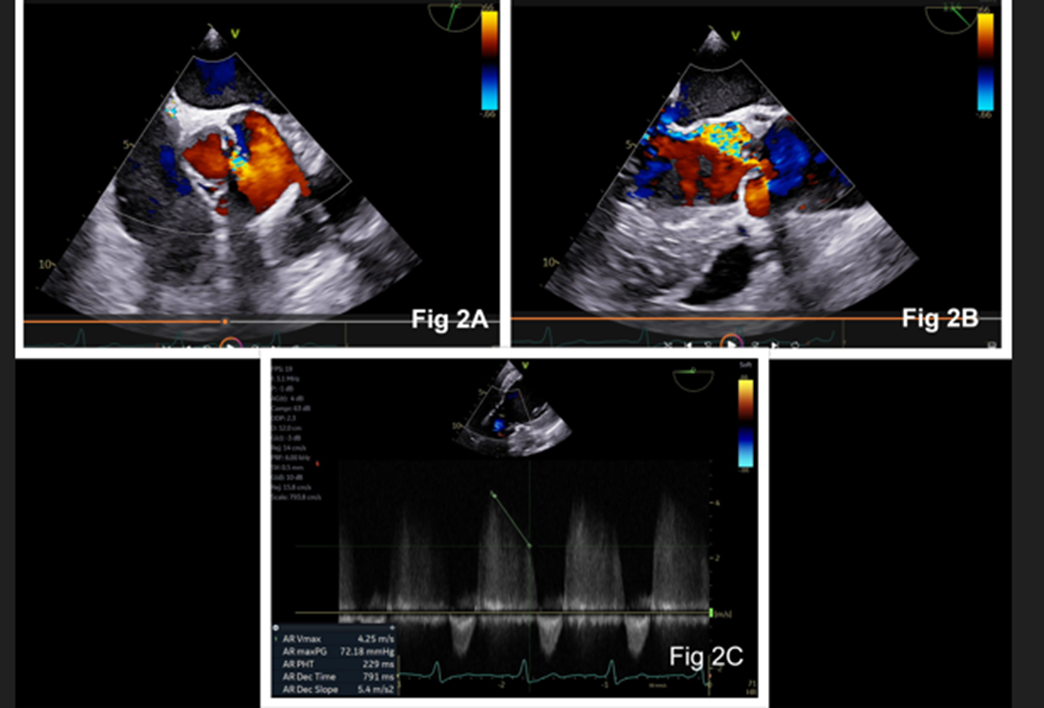
Figure 2: Two-dimensional echocardiography with color Doppler showing aortic regurgitation in aortic valve short-axis view (Figure 2A) and long-axis view (Figure 2B). Continuous wave Doppler at the aortic valve showing pressure half time and deceleration slope (Figure 2C)
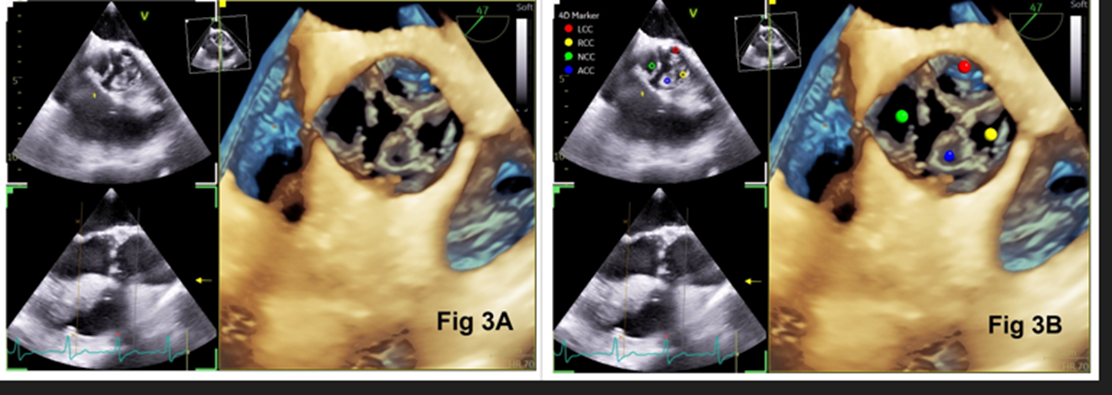
Figure 3: En-face three-dimensional view of the aortic valve showing 4 cusps (Figure 3A). A 4-D marker indicating left coronary cusp (red), right coronary cusp (yellow), non-coronary cusp (green), and accessory cusp (blue) [Figure 3B]
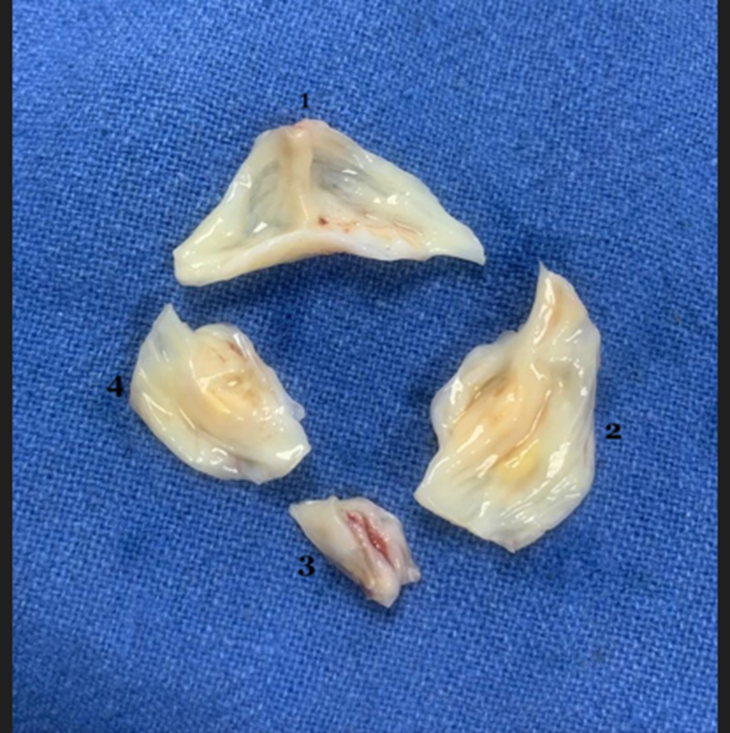
Figure 4: Photograph taken during surgery showing 3 large aortic cusps (1, 2, 4) representing left, right, and non-coronary cusps respectively, and one smaller cusp (3) representing accessory cusp
The QAV, first reported by Balinton in 1862, is a rare congenital anomaly with a prevalence of 0.008% to 0.043% in the autopsy and echocardiographic studies respectively. [1,5] The disease has a predilection for males with a mean presenting age between 45 to 60 years. Transthoracic echocardiography plays a significant role in detecting congenital anomalies of the aortic valve; however, it may be suboptimal for diagnosing QAV and associated malformations due to the poor acoustic window. Technological advances in echocardiography such as 3-D echocardiography and magnetic resonance imaging have enabled the increased diagnosis of this anomaly even in asymptomatic individuals. The characteristic echocardiographic finding in QAV is typical ‘cross’ or ‘X’ valve closure different from the characteristic ‘Y’ closure of the normal tricuspid aortic valve during diastole. During systole it appears as a rectangular opening, different from the typical triangular opening of the normal tricuspid aortic valve. 3-D echocardiography with the help of 4-D markers can correctly identify all four cusps.
Among the various hypotheses to explain the embryological basis of this anomaly, the most common is abnormal septation of the truncus arteriosus after septation of the arterial trunk. Abnormal cusp formation results from either aberrant fusion of the aorticopulmonary septum or from abnormal mesenchymal proliferation in the common trunk. The development of the aortic valve leaflets occurs immediately after the development of the coronary artery origins from the sinuses of Valsalva. It is therefore possible that anomalies in these two areas are related embryologically. Normally, three mesenchymal swellings develop into semilunar leaflets of the aortic and pulmonary trunks. In QAV, however, the fourth cusp arises during the early stage of truncal septation, resulting in either a different number of primordial aortic leaflets or abnormal cusp proliferation.6 The proposed mechanism of aortic regurgitation includes age‑related progressive leaflet fibrosis, unequal distribution of stress, and abnormal leaflet coaptation. The symptoms appear with an increase in the severity of aortic regurgitation and the development of left ventricular dysfunction. Aortic valve replacement for a QAV is generally the treatment of choice for patients with aortic regurgitation, with few cases of aortic valve repair reported. Naito et al have described aortic valve repair performed by suturing together the commissure between the right coronary cusp and the accessory cusp, thus converting the quadricuspid valve into a tricuspid valve.7 These patients are at risk of coronary ostial obstruction or injury at the time of valve replacement due to the abnormal origin and distribution of coronary arteries.8
In conclusion, QAV is a rare congenital anomaly and the majority of these patients will require surgery for aortic regurgitation. An early diagnosis and a regular follow-up may be helpful for the timely surgical intervention before ventricular decompensation occurs.
Clearly Auctoresonline and particularly Psychology and Mental Health Care Journal is dedicated to improving health care services for individuals and populations. The editorial boards' ability to efficiently recognize and share the global importance of health literacy with a variety of stakeholders. Auctoresonline publishing platform can be used to facilitate of optimal client-based services and should be added to health care professionals' repertoire of evidence-based health care resources.