
Case Report | DOI: https://doi.org/10.31579/2643-1912/008
*Corresponding Author: G Obi, Center for Cell and Gene Therapy, Baylor College of Medicine and Houston Methodist Hospital,Houston, TX, 77030, USA.
Citation: G Obi , K Dell Aquilla, S Liu , A Ewton, RT Kamble. (2020) Dermatopathic Lymphaden itis in Ruxolitinib Treated Graft Versus Host Disease..Stem cell Research and Therapeutics International. 2(1)
Copyright: © 2020 G Obi, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 26 November 2019 | Accepted: 23 December 2019 | Published: 10 January 2020
Keywords: dermatopathic lymphadenopathy; chronic graft versus host disease; Ruxolitinib
Dermatopathic Lymphadenopathy (DL) is a non-infectious, non-malignant, inflammatory lymphnode enlargement that is secondary to a cutaneous disorder [1]. It is most commonly reported in patients with cutaneous T cell lymphoma with axillary and inguinal lymphonodes as favored sites. In patients receiving allogeneic hematopoietic stem cell transplantation, an enlarged lymph node often signifies a disease relapse, post-transplant lymphoproliferative disorder (PTLD) or infection. We herein document DL in a patient treated with Ruxolitinib for cutaneous chronic graft versus host disease (cGvHD).
Dermatopathic Lymphadenopathy (DL) is a non-infectious, non-malignant, inflammatory lymphnode enlargement that is secondary to a cutaneous disorder [1]. It is most commonly reported in patients with cutaneous T cell lymphoma with axillary and inguinal lymphonodes as favored sites. In patients receiving allogeneic hematopoietic stem cell transplantation, an enlarged lymph node often signifies a disease relapse, post-transplant lymphoproliferative disorder (PTLD) or infection. We herein document DL in a patient treated with Ruxolitinib for cutaneous chronic graft versus host disease (cGvHD).
A 53 year old female developed right breast cancer (T2, N0, and M0) that was positive for estrogen receptor, progesterone receptor and HER2 in 2014. She underwent neoadjuvant and adjuvant chemotherapy with Pertuzumab, Herceptin, Docetaxel and Carboplatin (no anthracyclines). She was found to have germline mutation of p53 tumor suppressor gene and opted for contralateral mastectomy and bilateral salpingo-oophorectomy. Her only son tested negative for p53 mutation. In July 2017, while on adjuvant Tamoxifen, she developed pancytopenia; a bone marrow biopsy confirmed diagnosis of B cell acute lymphoblastic leukemia (ALL) that was negative for Philadelphia chromosome. She received induction with Hyper- CVAD along with prophylactic intrathecal treatments achieving complete remission.
In December 2017 she received an allogeneic hematopoietic stem cell transplantation (allo-HCT) from a mismatched unrelated donor (MMUD, 9/10, C mismatch) using Fludarabine, Melphalan, Alemtuzumab as conditioning; tacrolimus was used for GvHD prophylaxis. Unfortunately she received extremely low cell dose of 0.9 x 10 6/kg CD34+ cells resulting is engraftment (neutrophils day 16, platelets day 28) that was not sustained. Even with donor chimerism of 100%, the bone marrow cellularity was merely 10%. She required G-CSF support, red cells and platelets transfusion for next 6 months. She did not develop GvHD and remained in remission for ALL.
In July 2018, she required a second allogeneic transplant for graft failure receiving graft from a mismatched unrelated donor (MMUD, 9/10, mismatch at DQ). She received conditioning with Fludarabine, Melphalan and Alemtuzumab with tacrolimus as GvHD prophylaxis. Engraftment (neutrophil =day 15, platelets = 15) was followed by grade IV acute GvHD (grade III skin, grade II gut) and CMV reactivation necessitating hospitalization, treatment with systemic steroids and Ganciclovir/Letermovir. While aGvHD responded well to steroids, attempts to taper steroids less than 20 mg daily was associated with aGvHD exacerbation that persisted beyond day +100 as cutaneous cGvHD (Skin score -3, NIH overall 3). For steroid refractory cGvHD, she initiated Ruxolitinib (Jakafi, Incyte, Wilmington, DE) at 10 mg BID orally on day +232. Two months after introduction of Jakafi, steroid taper was initiated; after 3 months, Ruxolitinib dose was reduced to 10 mg daily. She currently has excellent control of her cGvHD with Ruxolitinib at 10 mg daily and prednisone 10 mg daily. On day + 310 of allo-HCT she reported enlarged left inguinal lymph node that was 3 x 2 cm in size; an additional right axillary lymph node was discovered on physical examination. She had no fever, weight loss or night sweats and her blood Epstein Barr Virus (EBV) polymerase chain reaction (PCR) was negative. A positron emission tomography (PET) scan identified right axillary lymphnode (1.2 x 1.1 cm, SUV 2.4) and left inguinal lymphnode (2 cm x 1.3 cm, SUV 3.9). A fine needle aspiration of lymph node was inconclusive. Excision biopsy of left inguinal lymph node showed marked expansion of paracortical Langerhans cells associated with variable pigment deposition without significant associated eosinophils (Figure-1).
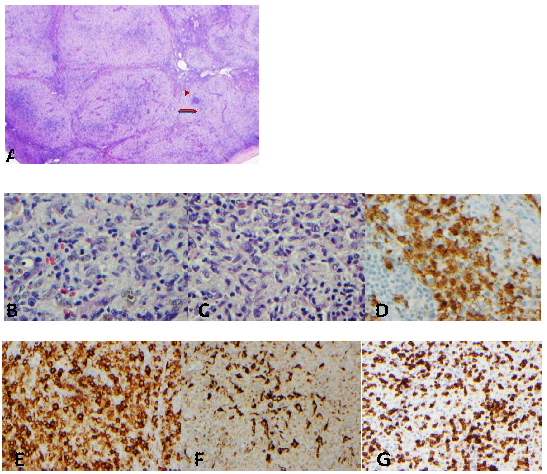
A: Marked paracortical expansion is noted. The lymphoid follicles are compressed. (HE x20). B &C: The expanded paracortical zones are composed of reactive T- cells, macrophages (some containing melanin) and Langerhans cells. (HE x400). D &E: S100 and CD1a immunostains highlight Langerhans cells. F: CD68 immunostain highlights macrophages. G: CD3 immunostain highlights T lymphocytes. (D: x400, E-G: x200)
There were areas of residual lymph node tissue. Immunohistochemical stains showed increased CD4 positive T cells showing no loss of pan T cell antigens CD2, CD5, CD7, or CD3. Immunohistochemical stain for CD68 showed increased histiocytes and CD1a and S100 highlighted numerous Langerhans cells. (Figure -1)Further chimeric characterization of the lymph node tissue confirmed all involved cells to be of donor origin.
Lymph node enlargement is not a common manifestation of cGvHD per se. When present, it generally signifies development of another pathology such as infection or a neoplastic process including PTLD. Patients with p53 germ line mutations develop early-onset sarcoma, breast cancer, adrenal cancer, brain tumor, acute leukemia and are susceptible to develop a second primary malignancy (SPM) after allo-HCT. We therefore chose to utilize non TBI preparative regimen that was reduced intensity in lieu of a fully myeloablative conditioning to reduce late SPM [2]. Our initial consideration was lymph node enlargement due to Ruxolitinib induced non-Hodgkin lymphoma as 16 fold increased non-Hodgkin lymphoma has been reported in patient with myelofibrosis (MF) receiving Ruxolitinib accounting for an incidence of 5.8% [3]. Reduction in T cell function and T-regulatory cells is implicated in the development of lymphoma that arises from a preexisting marrow clone in patients with MF. Such clones have been documented in 16% MF patients. While awaiting biopsy results we also considered Ruxolitinib associated infections including mycobacterial infections[4]. Our findings are similar to Kreft et al[5] reporting 2 cases of GvHD related DL documenting Langerhans’s cell (LC) chimerism in both. Following allo-HCT host antigen presenting cells are first to be replaced by donor cells; host LC however can persist for an extended period[6]. In our patient 100% cells in lymph node were of donor origin. As Ruxolitinib is being studied for treatment of Hemophagocytic lymphohistiocytosis, it seems unlikely that it may have contributed to development of DL. We believe our patient developed cutaneous cGvHD related DL even though DL developed after improved but persisting cGvHD.
Although rare, DL mediated by donor LC in allo-HCT is easily recognizable on histology facilitating patient counseling and management.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
