AUCTORES
Globalize your Research

case report | DOI: https://doi.org/10.31579/2690-4861/737
1Department of Gynecology and Obstetrics, The First Affiliated Hospital of JiNan University, 613 Huangpu Road West, 510630 Guangzhou, People’s Republic of China.
2Department of Pediatrics, Guangzhou, Nansha District maternal and child health hospital, 511466, Guangzhou, People’s Republic of China.
*Corresponding Author: Shanrong Shu, Department of Gynecology and Obstetrics, The First Affiliated Hospital of JiNan University, 613 Huangpu Road West, 510630 Guangzhou, People’s Republic of China.
Citation: Qi Chen, Liping Lai, Li Li, Fanyuan Li, Shanrong Shu, (2025), Chromosomal Microarray Analysis Detection of Microdeletion on Chromosome 17 in Fetus, International Journal of Clinical Case Reports and Reviews, 24(4); DOI:10.31579/2690-4861/737
Copyright: Qi Chen, Liping Lai, Li Li, Fanyuan Li, Shanrong Shu, (2025), Chromosomal Microarray Analysis Detection of Microdeletion on Chromosome 17 in Fetus, International Journal of Clinical Case Reports and Reviews, 24(4); DOI:10.31579/2690-4861/737
Received: 11 March 2025 | Accepted: 19 March 2025 | Published: 26 March 2025
Keywords: fetal deformity; microarray analysis; karyotype analysis
Backgroud:17p12microdeletion syndrome is a relatively rare chromosomal abnormality, which was accompanied with phenotypic variability. The reports on prenatal ultrasound abnormalities of fetus with 17p12 microdeletion are rare. Here we reported a case found by abnormal ultrasound appearance and verified by chromosomal microarray analysis.
Methods: Ultrasoundgraphy in the first and second trimester of pregnancy was adopted to detect the development of organs. Chromosomal karyotype analysis was taken to analyze the number and structure of chromosome. Microdeletion of about 1.4Mb in the short arm of chromosome 17p12 were detected by chromosomal microarray analysis and were confirmed by copy number variation analysis based on high-throughput sequencing technology.
Results: Chromosomal microarray analysis showed arr [hg19]17p12 (14, 087,919-15, 479, 940)×1, with microdeletion size of 1.4Mb, which involved COX10(602125), PMP (601097) and other five OMIN genes. Ultrasound manifested multiple deformity during routine prenatal examination. The karyotype analysis showed no abnormalities.
Conclusion: The microdeletion fragment of fetal chromosome 17 was successfully detected and diagnosed. When fetal malformations are detected by ultrasound, but no abnormalities are found in karyotype analysis, further microarray analysis is required.
It is estimated that human have ∼22,000 genes directing protein synthesis, which are necessary for maintaining structure and function for our body[1]. If there are serious errors in those genes during the embryonic stage, it will induce congenital diseases [2] or fetal deformity [3] and even fetal death [4]. Trisomy 21 (Down's syndrome), trisomy 13 (Patau syndrome), trisomy 18 (Edward's syndrome), and sex chromosome aneuploidy are the most common chromosomal aneuploidy[5,6], which can be detected by routine karyotype analysis.
Chromosomal microarray analysis is using array comparative genomic hybridization or a single nucleotide polymorphism array to explicitly understand the structure of genome [7],which was widely adopted to detect the underlying mechanism for fetal deformity.
Here, we reported a prenatal case with 17p12 microdeletion presenting with abnormal ultrasound findings. Meanwhile, we also made a review on prenatal cases involving similar 17p12 deletion.
A 28-year-old pregnant woman, pregnancy 1, parturition 0, gestational age 16+1 weeks, was sent to the First Affiliated Hospital of JiNan University. The woman was 161 cm tall and weighed 62kg. Ultrasonography at 13 weeks of gestational age had revealed nasal bone missing, asymmetric bilateral choroid plexus, deviated midbrain midline, flattened face and widen distance of binocular. At the 16+1 weeks of gestational age, the above mentioned deformity became more obvious shown by ultrasonography. The serological screening for the risk of 18/13/21-trisomy syndrome and fetal growth restriction was 1:35/1:215/1:8 and 1:78 respectively. Amniotic fluid was taken for prenatal diagnosis, including karyotype analysis and chromosomal microarray analysis (CMA). The CMA results showed that there was about1.4 Mb of microdeletions in 17p12, which involved COX10(602125), PMP (601097) and other five OMIN gene, while the karyotype analysis showed no significant abnormalities.
Material and Methods
Chromosome karyotype analysis
With the high risk for trisomy 21, we suggested the patient to experience amniocentesis to verify the fetal karyotype analysis. The amniocentesis was performed under the guidance of ultrasound graphy, and15-20 ml of amniotic fluid was taken. The karyotype analysis of fetal amniotic fluid exfoliated cells was performed by visual identification.
Chromosomal microarray analysis
CMA was performed on 10ml uncultured amino fluid cells and 5ml peripheral blood cells according to the manufacturers protocol. DNA extraction, sequencing library preparation, sequencing, and other processes were performed as described. Sequencing was performed using an Ion Proton Sequencing System (Life Technologies). The results were analyzed using the chromosome analysis software.
Amniotic fluid karyotype analysis showed no obvious abnormalities in fetal chromosome structure (Figure.1a), and the result of CMA detected 1.4Mb microdeletion in17p12, showed as arr [hg19] 17p12 (14, 087, 919-15, 479, 940)×1 (Figure.1b). Though CMA demonstrated the microdeletion, considering the multiple abnormality in the appearance, we still recommended the family take exon sequencing, which was performed by another company. No gene mutation was found in the parents. But in the fetus, the CCDC88C gene associated with spinocerebellar ataxia type 40, also known as congenital hydrocephalus type 1, was detected to be mutated. And beyond that, two unspecified variants were detected in the KIAA1109 gene, which was associated with Alkuraya-Kucinskas syndrome. Lastly, the pregnant women experienced induce of labor by in injection of drug to the amniotic cavity and the multiple deformities were testified (shown in Figure.1c).
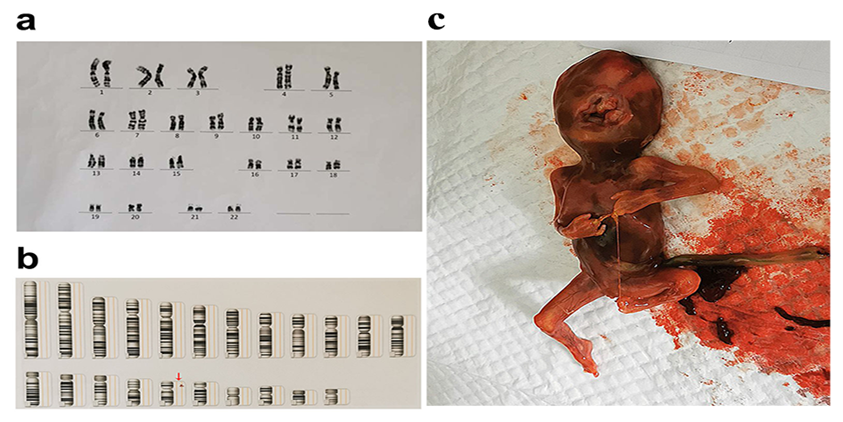
Figure 1: The results of chromosomal karyotype analysis, chromosomal microarray analysis and the discharged fetus. a. the normal chromosomal karyotype analysis with unreported sex chromosomes. b. chromosomal microarray analysis showed the abnormal gene arr[hg19] 17p12(14, 087, 919-15, 479, 940)×1 demonstrated by the red arrow. c. the discharged fetus testified multiple deformities
In clinical practice, birth defects mainly result from fetal chromosomal abnormality, and over 95% of chromosomal abnormality is aneuploidies, such as trisomy 21, trisomy 18, trisomy 13, and sex chromosome abnormalities, which can be examined by chromosomal karyotype analysis[8]. With the development of detection technology, we find that some chromosome abnormalities are due to microdeletions or duplications. At present, the detection techniques for those microdeletions or duplication mainly depended on FISH and CMA.
CMA is a method to detect the increment or loss of DNA in the whole genome. It can detect chromosome aneuploidy and large fragments of chromosome structure changes, such as those which are too small to be detected by traditional methods. It is reported that chromosome deficiencies and duplications of less than 5 Mb by amniotic fluid karyotype analysis is difficult and they may often be missed [9]. With the limitation of chromosomal karyotype analysis, we did not find abnormalities in the first stage.
Most children with chromosomal microdeletions can survive normally, but they may exhibit various degrees of physical or mental abnormalities after birth [10]. Also, we found many fatal deformities including appearance and organ development, which could be significantly detected by ultrasoundgraphy. In view of eugenics, the pregnant woman chose to terminate the pregnancy.
The results of the CMA inthe study showed that there was a microdeletion of 1.4Mb in the position of p12of autosomal chromosome 17. The deletion of chromosome 17 will result to Smith-Magenis syndrome [11], including the pathogenic gene PMP22 for recurrent transient pressure palsies [12], paranoid-hallucinatory schizophrenia[13] and malignant mesothelioma [14]. Smith-Magenis syndrome (SMS; OMIM #182290) is a complex genetic disorder characterized by distinctive physical features, development delay, cognitive impairment, and a typical behavioral phenotype[15]. In our case, the fetus demonstrated the obvious deformities similar to the report. SMS is caused by interstitial 17p12 deletions, encompassing multiple genes and including the retinoic acid-induced 1 gene (RAI1), or by mutations in RAI1 itself [16]. The results of exon sequencing demonstrated a novel mutation in CCDC88C gene and KIAA1109, which is responsible for familial ataxia, tremor, dementia [17] and severe disorder of brain development [18]. In the case presented here, the microdeletion mutation in the17p12 region cause a multiple deformity, which is according to the reported literatures. We examined the chromosome of the parents, we did not something abnormal. The microdeletion in17p12 in the fetus may come from abnormal pairing of sperm and egg. Fortunately, the abnormalities resulted to obvious abnormal appearance, which can be examined by ultrasoundgraphy.
From our case, we concluded that ultrasound examination is a necessary examination during childbirth. Although the chromosome karyotype analysis is normal, chromosome microdeletion or displacement may still exist. We need further examination to explore the mechanism leading to fetus abnormalities.
None
The study was approved by an ethics committee of JiNan University. We have obtained informed consent during the treatment and got consent to publish the case from the study participant.
There were no competing interests among authors
This project was supported by the National College Students Innovation and Entrepreneurship Training Program (CX18024) and Science and Technology Projects in Guangzhou (2021 02010134)
The data was available when necessary
Clearly Auctoresonline and particularly Psychology and Mental Health Care Journal is dedicated to improving health care services for individuals and populations. The editorial boards' ability to efficiently recognize and share the global importance of health literacy with a variety of stakeholders. Auctoresonline publishing platform can be used to facilitate of optimal client-based services and should be added to health care professionals' repertoire of evidence-based health care resources.